- Главная
- Разное
- Дизайн
- Бизнес и предпринимательство
- Аналитика
- Образование
- Развлечения
- Красота и здоровье
- Финансы
- Государство
- Путешествия
- Спорт
- Недвижимость
- Армия
- Графика
- Культурология
- Еда и кулинария
- Лингвистика
- Английский язык
- Астрономия
- Алгебра
- Биология
- География
- Детские презентации
- Информатика
- История
- Литература
- Маркетинг
- Математика
- Медицина
- Менеджмент
- Музыка
- МХК
- Немецкий язык
- ОБЖ
- Обществознание
- Окружающий мир
- Педагогика
- Русский язык
- Технология
- Физика
- Философия
- Химия
- Шаблоны, картинки для презентаций
- Экология
- Экономика
- Юриспруденция
Моногенные болезни презентация
Содержание
- 1. Моногенные болезни
- 2. Моногенные болезни (МБ) — это заболевания, в
- 3. В настоящее время известно более 6000 нозологических
- 5. НУКЛЕОТИД ДНК
- 6. А а Аллельные гены –
- 7. Классификация МБ I. По частоте встречаемости:
- 8. II. Основная патогенетическая классификация НБО (НДО)
- 9. Дефект гена определяет дефект белка-фермента в
- 10. ФЕНИЛКЕТОНУРИЯ (ФКУ) ФЕНИЛАЛАНИН
- 11. ФКУ
- 12. Болезнь манифестирует в возрасте 2-6 месяцев.
- 13. 2. Моногенные синдромы ВПР -
- 14. Синдром МАРФАНА – наследственное заболевание, связанное
- 15. С-м Марфана Нарушение
- 16. - астенический тип телосложения; - дефицит
- 17. С-м Марфана у девочки 14 лет
- 19. С-м Марфана арахнодактилия
- 20. С-м Марфана арахнодактилия
- 21. Долихостеномелия. Арахнодактилия. Положительный симптом запястья.
- 22. Симптом «большого пальца» при арахнодактилии.
- 23. С-мы большого пальца и запястья
- 24. Марфана с-м – подвывих хрусталика
- 25. IQ - N
- 26. III. По типу мутации: Миссенс –
- 27. С-М МАРТИН-БЭЛЛ- СИНДРОМ УМСТВЕННОЙ ОТСТАЛОСТИ С
- 28. Характерный фенотип: - прямоугольное лицо; - большие
- 29. IV. По типу наследования: Аутосомно-доминантный Аутосомно-рецессивный Х-сцепленный доминантный Х-сцепленный рецессивный Y-сцепленный Митохондриальный
- 30. Аутосомно-доминантный тип наследования
- 31. Аутосомно-доминантным называется заболевание, развитие которого обусловлено доминантным
- 32. Характерны два типа родословных: - при
- 33. Вертикальный тип наследования Поражение лиц обоих полов
- 34. Единичный случай рождения больного ребенка в здоровой
- 35. При неполной пенетрантности гена – в родословной имеются «проскакивающие» поколения
- 36. Аутосомно-рецессивный тип наследования
- 37. Аутосомно-рецессивным называется заболевание, развитие которого обусловлено рецессивным
- 38. Горизонтальный тип наследования Поражение лиц обоих полов Последующий риск – 25%
- 39. «Выщеплению» гомозигот способствует инбридинг - кровнородственный брак
- 40. Сцепленными с полом называются заболевания,
- 41. Х-сцепленный рецессивный тип наследования (заболевание вызывается рецессивным ген, локализованным в негомологичном участке Х хромосомы)
- 42. Поражение лиц мужского пола Матери – носительницы патологического гена (Х*) ½ ¼
- 43. Особенности клиники МБ I. Широкий
- 44. ПОЛИАЛЛЕЛИЗМ (множественный аллелизм) явление, при котором
- 45. ПОЛИЛОКУСНОСТЬ явление, при котором за синтез
- 46. В настоящее время различают: Классическую ФКУ (I
- 47. различная комбинация генов-модификаторов различная доза патологического
- 48. ГЕНОМНЫЙ ИМПРИНТИНГ – с-м Прадера-Вилли Причина заболевания
- 49. ГЕНОМНЫЙ ИМПРИНТИНГ – с-м Ангельмана Причина
- 50. 2. Варьирующий возраст начала заболевания Внутриутробно реализуется
- 51. 3. Неодновременность проявления признаков Например,
- 52. 4. Наличие у больного редко встречающихся специфических
- 53. С-м Беквита-Видемана Макросомия Макроглоссия Пупочная грыжа Насечки на мочке уха
- 54. МУКОВИСЦИДОЗ Муковисцидоз или кистофиброз — это патология
- 55. Ген болезни локализован в 7q31.1-32 и кодирует
- 56. Основной патогенетический механизм болезни – увеличение вязкости
- 57. Меконеальный илеус Кишечная непроходимость у новорожденного
- 58. Легочная форма Закупорка просвета мелких респираторных путей
- 59. Бронхи человека, больного муковисцидозом. Гиперпродукция слизи приводит
- 60. Кишечная форма МВ Изменение водно-электролитного состава панкреатического
- 61. Клиническая картина кишечной формы муковисцидоза обусловлена недостаточностью ферментативной
- 62. Смешанная форма МВ
- 63. Неонатальный скрининг – обследование всех новорожденных
- 64. Неонатальный скрининг Забор крови на 5 – 7 сутки жизни
- 65. ФЕНИЛКЕТОНУРИЯ ВРОЖДЕННЫЙ ГИПОТИРЕОЗ ГАЛАКТОЗЕМИЯ АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ МУКОВИЗЦИДОЗ
- 66. Спасибо за внимание!

Слайд 2 Моногенные болезни (МБ) — это заболевания, в основе которых лежит мутация
одного гена, в результате чего нарушается или выключается полностью функция соответствующего белка.
Известно > 7 тыс. МБ
У 2,4 % населения
Частота 10:1000
Известно > 7 тыс. МБ
У 2,4 % населения
Частота 10:1000
 — это заболевания, в основе которых лежит мутация одного гена, в результате")
Слайд 3 В настоящее время известно более 6000 нозологических единиц МБ.
На 1000
новорожденных МБ выявляются у 42 — 65 детей
(4,2 — 6,5%).
В структуре общей смертности детей до 5 лет на долю МБ приходится 8 — 10%.
(4,2 — 6,5%).
В структуре общей смертности детей до 5 лет на долю МБ приходится 8 — 10%.

Слайд 4
Ген – участок молекулы ДНК, выполняющий определенную функцию
Геном – совокупность всех
генов организма (всей ДНК соматической клетки)
Генотип – совокупность генов соматической клетки (организма), которая проявляется фенотипически
Фенотип – совокупность всех свойств и признаков организма, которая формируется под влиянием генотипа и факторов внешней среды
Генотип – совокупность генов соматической клетки (организма), которая проявляется фенотипически
Фенотип – совокупность всех свойств и признаков организма, которая формируется под влиянием генотипа и факторов внешней среды


Слайд 6А
а
Аллельные гены –
гены, расположенные в гомологичных локусах гомологичных хромосом и
отвечающие за развитие альтернативных свойств одного признака
Генотип м.б. гомо- или гетерозиготным
Генотип м.б. гомо- или гетерозиготным

Слайд 7Классификация МБ
I. По частоте встречаемости:
1. Часто встречающиеся
1
: 10 тыс. новорожденных и чаще
(ФКУ, муковисцидоз, синдром Мартина-Бэлл)
2. Редко встречающиеся
1 : 100 тыс. и реже
3. Со средней частотой встречаемости
1 : 10 - 100 тыс. новорожденных
(ФКУ, муковисцидоз, синдром Мартина-Бэлл)
2. Редко встречающиеся
1 : 100 тыс. и реже
3. Со средней частотой встречаемости
1 : 10 - 100 тыс. новорожденных

Слайд 8II. Основная патогенетическая классификация
НБО (НДО) – наследственные болезни обмена (ферментопатии)
НБО аминокислот
(фенилкетонурия)
НБО углеводов (галактоземия)
НБО липидов (гиперлипидемия, гиперхолестеринемия)
НБО пуринов и пиримидинов (подагра)
НБ биосинтеза кортикостероидов (адреногенитальный с-м)
НБ порфиринового и билирубинового обмена (порфирии)
НБО металлов (болезни Вильсона-Коновалова)
НБ эритрона (гемолитические анемии)
НБ лимфоцитов и лейкоцитов (септический грануломатоз)
НБ транспорта систем почек (вит. D-резистентный рахит)
НБО углеводов (галактоземия)
НБО липидов (гиперлипидемия, гиперхолестеринемия)
НБО пуринов и пиримидинов (подагра)
НБ биосинтеза кортикостероидов (адреногенитальный с-м)
НБ порфиринового и билирубинового обмена (порфирии)
НБО металлов (болезни Вильсона-Коновалова)
НБ эритрона (гемолитические анемии)
НБ лимфоцитов и лейкоцитов (септический грануломатоз)
НБ транспорта систем почек (вит. D-резистентный рахит)
 – наследственные болезни обмена (ферментопатии)НБО аминокислот (фенилкетонурия)НБО углеводов (галактоземия)НБО")
Слайд 9Дефект гена определяет дефект белка-фермента в результате блокируется б/х реакция, количество субстрата
в клетке увеличивается,
количество продуктов реакции уменьшается
Избыток субстрата перерабатывается в побочные продукты метаболизма
СУБСТРАТ ПРОДУКТ
побочные метаболиты

Слайд 10ФЕНИЛКЕТОНУРИЯ (ФКУ)
ФЕНИЛАЛАНИН
ТИРОЗИН
фенилаланингидроксилаза
ГЕН
фенилпировиноградная кислота
фенилмолочная кислота
фенилуксусная кислота
фенилаланингидроксилаза
ГЕН
фенилпировиноградная кислота
фенилмолочная кислота
фенилуксусная кислота
ФЕНИЛАЛАНИН ТИРОЗИН фенилаланингидроксилаза")

Слайд 12Болезнь манифестирует в возрасте 2-6 месяцев.
Характерно:
вялость ребенка, отсутствие интереса
к окружающему;
иногда повышенная раздражительность, беспокойство, срыгивание;
нарушение мышечного тонуса (чаще мышечная гипотония);
судороги, признаки аллергического дерматита;
«мышиный» запах;
Постепенно формируется
задержка психомоторного развития;
микроцефалия;
эпилептические приступы;
умственная отсталость глубокой степени
(IQ - около 20 ед. при норме 85 - 115)
иногда повышенная раздражительность, беспокойство, срыгивание;
нарушение мышечного тонуса (чаще мышечная гипотония);
судороги, признаки аллергического дерматита;
«мышиный» запах;
Постепенно формируется
задержка психомоторного развития;
микроцефалия;
эпилептические приступы;
умственная отсталость глубокой степени
(IQ - около 20 ед. при норме 85 - 115)

Слайд 132. Моногенные
синдромы ВПР - синдромы врожденных пороков,
развитие
которых обусловлено мутацией одного гена

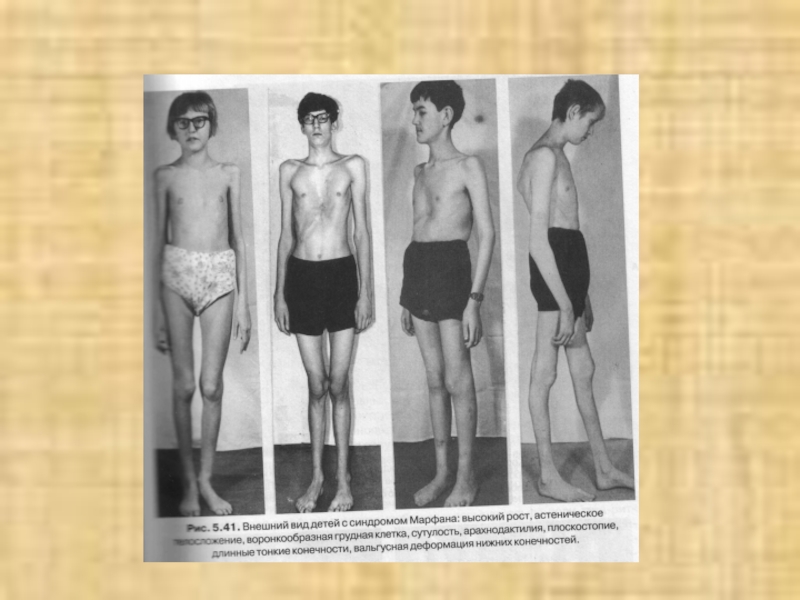
Слайд 14Синдром МАРФАНА –
наследственное заболевание, связанное с нарушениями обмена соединительной ткани.
Болезнь описана В. Марфаном в 1896 году.
Частота встречаемости 1: 10 000 – 15 000.
Развитие заболевания обусловлено мутацией в гене фибриллина, который локализован в длинном плече хромосомы 15 (локус 15q21).
Выявлено несколько типов мутаций гена (в основном миссенс).
Тип наследования – аутосомно-доминантный с высокой пенетрантностью гена.
Частота встречаемости 1: 10 000 – 15 000.
Развитие заболевания обусловлено мутацией в гене фибриллина, который локализован в длинном плече хромосомы 15 (локус 15q21).
Выявлено несколько типов мутаций гена (в основном миссенс).
Тип наследования – аутосомно-доминантный с высокой пенетрантностью гена.

Слайд 15
С-м Марфана
Нарушение
опорно- двигательного аппарата
Нарушение органа зрения -
подвывих хрусталика
Изменение сердечно-
сосудистой системы-
нарушение проводимости, формирование аневризмы аорты.
нарушение проводимости, формирование аневризмы аорты.

Слайд 16 - астенический тип телосложения; - дефицит массы тела; - долихостеномелия; - арахнодактилия; - искривление позвоночника; -
деформация грудной клетки









Слайд 26 III. По типу мутации: Миссенс – замена одного нуклеотида на другой. Нонсенс –
замена нуклеотидов, в результате которой происходит формирование стоп-кодона.
«Сдвиг рамки считывания» - при выпадении или вставке (инсерции) нуклеотида, что вследствие неперекрываемости генетического кода приводит к формированию новых триплетов.
Сплайсинговая мутация.
Экспансия тринуклеотидных повторов

Слайд 27С-М МАРТИН-БЭЛЛ-
СИНДРОМ УМСТВЕННОЙ ОТСТАЛОСТИ С ЛОМКОЙ
Х ХРОМОСОМОЙ
Ген FMR1 локализован
в Х хромосоме
В гене есть вариабельная область, в которой триплет ГЦЦ в норме повторяется от 6 до 42;
При увеличении числа триплетов (экспансии) до 200 – состояние премутации;
При увеличении числа триплетов свыше 200 – мутация, при которой развивается клиника заболевания
В гене есть вариабельная область, в которой триплет ГЦЦ в норме повторяется от 6 до 42;
При увеличении числа триплетов (экспансии) до 200 – состояние премутации;
При увеличении числа триплетов свыше 200 – мутация, при которой развивается клиника заболевания

Слайд 28Характерный фенотип:
- прямоугольное лицо;
- большие оттопыренные ушные раковины;
выступающий лоб;
массивный
подбородок;
макроорхизм;
умственная отсталость умеренная или глубокая;
аутизм
Тяжесть течения заболевания коррелирует с количеством повторов
макроорхизм;
умственная отсталость умеренная или глубокая;
аутизм
Тяжесть течения заболевания коррелирует с количеством повторов

Слайд 29
IV. По типу наследования:
Аутосомно-доминантный
Аутосомно-рецессивный
Х-сцепленный доминантный
Х-сцепленный рецессивный
Y-сцепленный
Митохондриальный


Слайд 31Аутосомно-доминантным называется заболевание, развитие которого обусловлено доминантным геном, который локализован в
аутосоме
Больной имеет генотип АА или Аа

Слайд 32Характерны два типа родословных: - при заболеваниях, при которых больной доживает до
репродуктивного возраста, вступает в брак и сохраняет фертильность;
- при тяжелейших заболеваниях, когда продолжительность жизни больного значительно сокращена и(или) больной бесплоден


Слайд 34 Единичный случай рождения больного ребенка в здоровой семье – следствие генеративной
генной доминантной мутации
Последующий риск – около 0%
Последующий риск – около 0%



Слайд 37Аутосомно-рецессивным называется заболевание, развитие которого обусловлено рецессивным геном, локализованным в аутосоме. Генотип
пациента - аа



Слайд 40 Сцепленными с полом называются заболевания, гены которых расположены в негомологичных
участках половых хромосом
Х Y

Слайд 41Х-сцепленный рецессивный тип наследования (заболевание вызывается рецессивным ген, локализованным в негомологичном участке
Х хромосомы)
")
½¼")
Слайд 43
Особенности клиники МБ
I. Широкий клинический полиморфизм, генетическими причинами которого являются:
- полиаллелизм;
-
полилокусность;
- различная комбинация генов-модификаторов;
- различная доза патологического гена;
- явление геномного импринтинга
- различная комбинация генов-модификаторов;
- различная доза патологического гена;
- явление геномного импринтинга

Слайд 44ПОЛИАЛЛЕЛИЗМ
(множественный аллелизм)
явление, при котором в генофонде популяции существует более двух
аллелей
Например, при ФКУ:
А – аллель, определяющий > 70% активности ф-та;
а1 – аллель, определяющий 30% активности ф-та;
а2 – аллель, определяющий 10% активности ф-та;
а3 – аллель, определяющий 0% активности ф-та
У пациента с генотипом а3а3 заболевание протекает более тяжело, чем у больного с генотипом а1а1
Например, при ФКУ:
А – аллель, определяющий > 70% активности ф-та;
а1 – аллель, определяющий 30% активности ф-та;
а2 – аллель, определяющий 10% активности ф-та;
а3 – аллель, определяющий 0% активности ф-та
У пациента с генотипом а3а3 заболевание протекает более тяжело, чем у больного с генотипом а1а1
 явление, при котором в генофонде популяции существует более двух аллелейНапример, при ФКУ:А")
Слайд 45ПОЛИЛОКУСНОСТЬ
явление, при котором за синтез белковой молекулы отвечает 2 и
более генов.
ГЕН 1 ГЕН 2
ГЕН 1 ГЕН 2
апофермент
кофермент

Слайд 46 В настоящее время различают:
Классическую ФКУ (I типа) - ген 12q22-q24.2
Атипичные формы
(ФКУ I – VII типов)
Атипичные формы ФКУ обусловлены недостаточностью тетрагидробиоптерина – кофактора гидролаз фенилаланина, тирозина и триптофана
гены 11q22.3-q23.3
14q22.1-q22.2
4p15.31
Атипичные формы ФКУ обусловлены недостаточностью тетрагидробиоптерина – кофактора гидролаз фенилаланина, тирозина и триптофана
гены 11q22.3-q23.3
14q22.1-q22.2
4p15.31
 - ген 12q22-q24.2Атипичные формы (ФКУ I – VII")
Слайд 47различная комбинация генов-модификаторов
различная доза патологического гена (АА или Аа)
явление геномного импринтинга
– различная активность гена в зависимости от родительского происхождения хромосомы, в которой локализован этот ген.
явление геномного импринтинга – различная активность гена")
Слайд 48ГЕНОМНЫЙ ИМПРИНТИНГ –
с-м Прадера-Вилли
Причина заболевания – инактивация генов q11-13 хромосомы 15
отцовского происхождения
- ожирение
олигофрения
гипогонадизм
- ожирение
олигофрения
гипогонадизм

Слайд 49ГЕНОМНЫЙ ИМПРИНТИНГ –
с-м Ангельмана
Причина заболевания
инактивация генов
хромосомы 15
материнского происхождения
олигофрения
приступы судорог, резкие
движения (особенно рукоплескания)
частый беспричинный смех или улыбка
частый беспричинный смех или улыбка
частый")
Слайд 502. Варьирующий возраст начала заболевания
Внутриутробно реализуется 25% генных мутаций;
До начала пубертата
– 45%;
В течение пубертатного периода – 20%;
После 20 лет – 10%.
В течение пубертатного периода – 20%;
После 20 лет – 10%.

Слайд 513. Неодновременность проявления признаков
Например, при синдроме Марфана:
при рождении диагностируется
арахнодактилия
к 3 годам – патология зрения
к 7 – 8 годам - патология ССС.
к 3 годам – патология зрения
к 7 – 8 годам - патология ССС.

Слайд 524. Наличие у больного редко встречающихся специфических симптомов.
Например,
вертикальные насечки на мочке уха при синдроме Беквита-Видеманна


Слайд 54МУКОВИСЦИДОЗ
Муковисцидоз или кистофиброз — это патология экзокринных желез (бронхиальных, потовых, слезных,
слюнных), а также поджелудочной железы и печени, проявляющаяся выделением секрета повышенной вязкости и сопровождающаяся вторичными изменениями в легких, поджелудочной железе и кишечнике
, а также поджелудочной")
Слайд 55 Ген болезни локализован в 7q31.1-32 и кодирует белок-регулятор трансмембранной проводимости для
ионов хлора
(CFTR — кистофиброзный трансмембранный регулятор) А-Р тип наследования
(CFTR — кистофиброзный трансмембранный регулятор) А-Р тип наследования

Слайд 56Основной патогенетический механизм болезни – увеличение вязкости секрета, выделяемого слизеобразующими железами
бронхов, кишечника, поджелудочной железы, семенников и придаточных пазух носа, что приводит к их закупорке.
Клинические формы:
Меконеальный илеус (3%)
Легочная (15-20%)
Кишечная (10%)
Смешанная (75-80%)
Клинические формы:
Меконеальный илеус (3%)
Легочная (15-20%)
Кишечная (10%)
Смешанная (75-80%)

Слайд 57Меконеальный илеус
Кишечная непроходимость у новорожденного
Рентгенологическое обследование новорожденного с мекониальной непроходимостью.

Слайд 58Легочная форма
Закупорка просвета мелких респираторных путей
Присоединение вторичной инфекции
Хронический воспалительный процесс в
бронхо-легочной системе: бронхиты, пневмонии, абсцессы, бронхоэктазы

Слайд 59 Бронхи человека, больного муковисцидозом. Гиперпродукция слизи приводит к тому, что заполненные
ею бронхи затрудняют дыхание и служат пристанищем для многих болезнетворных бактерий. (Фото CNRI.)

Слайд 60Кишечная форма МВ
Изменение водно-электролитного состава панкреатического сока, его сгущение и затруднение
выделения в просвет кишечника
Нарушение функции кишечника, нарушение формирования каловых масс, непроходимость кишечника
Кистозно-фиброзное изменение ткани поджелудочной железы
Нарушение функции кишечника, нарушение формирования каловых масс, непроходимость кишечника
Кистозно-фиброзное изменение ткани поджелудочной железы

Слайд 61 Клиническая картина кишечной формы муковисцидоза обусловлена недостаточностью ферментативной активности желудочно-кишечного тракта, которая
особенно ярко проявляется после перевода ребенка на искусственное вскармливание или прикорм. Расщепление и всасывание питательных веществ снижено, в кишечнике преобладают гнилостные процессы, сопровождающиеся накоплением газов. Очень частый стул, суточный объём каловых масс в 28 раз может превышать возрастную норму. Вздутие живота становится причиной схваткообразных болей животе.


Слайд 63 Неонатальный скрининг –
обследование всех новорожденных с целью раннего (доклинического) выявления
МБ
Показания для проведения скрининга:
Высокая частота встречаемости МБ
Принципиальная необходимость раннего начала лечения
Разработанные методы лечения
Показания для проведения скрининга:
Высокая частота встречаемости МБ
Принципиальная необходимость раннего начала лечения
Разработанные методы лечения
 выявления МБПоказания для проведения скрининга:Высокая")








