- Главная
- Разное
- Дизайн
- Бизнес и предпринимательство
- Аналитика
- Образование
- Развлечения
- Красота и здоровье
- Финансы
- Государство
- Путешествия
- Спорт
- Недвижимость
- Армия
- Графика
- Культурология
- Еда и кулинария
- Лингвистика
- Английский язык
- Астрономия
- Алгебра
- Биология
- География
- Детские презентации
- Информатика
- История
- Литература
- Маркетинг
- Математика
- Медицина
- Менеджмент
- Музыка
- МХК
- Немецкий язык
- ОБЖ
- Обществознание
- Окружающий мир
- Педагогика
- Русский язык
- Технология
- Физика
- Философия
- Химия
- Шаблоны, картинки для презентаций
- Экология
- Экономика
- Юриспруденция
Наследственные иммунодефициты презентация
Содержание
- 1. Наследственные иммунодефициты
- 2. Наследственные иммунодефицитные состояния — это гетерогенная группа
- 3. Клинические проявления иммунодефицитных состояний обусловлены снижением активности
- 4. Распространенность первичных наследственных иммунодефицитов среди населения существенно
- 5. Недостаточность системы В-лимфоцитов и гуморального звена
- 6. В зависимости от преимущественного повреждения клеток иммунной системы все иммунодефициты разделены на 4 группы
- 7. (1) Т-зависимые, клеточные с повреждением клеточного иммунитета,
- 8. Важнейшая роль в производстве антител принадлежит генам
- 9. В ходе созревания лимфоцитов от стволовой клетки
- 10. Цитокиновая система обеспечивает взаимодействие между лимфоцитами и
- 11. В основе развития наследственных иммунодефицитов могут
- 12. При наиболее тяжелых формах комбинированного иммунодефицита нарушается
- 13. При хроническом гранулематозе наблюдается недостаточность
- 14. Экспрессия генов иммуноглобулинов и Т-клеточных рецепторов
- 15. Организм каждого человека может вырабатывать около 1011
- 16. Разнообразие генов иммуноглобулинов, контролирующих синтез антител, достигается
- 17. Изначально антитела кодируются относительно небольшим количеством генов,
- 18. Эта перестройка сопровождается резким увеличением частоты соматических
- 19. Молекулы иммуноглобулинов формируются из двух тяжелых (H)
- 20. Постоянный участок определяет класс иммуноглобулинов (M,
- 21. Аминокислотная последовательность V-участков очень изменчива у разных
- 22. В половых клетках человека нет полноразмерных генов,
- 23. Так, локус для V-участка H-цепи состоит из
- 24. Причем гены вариабельного участка перемежаются с генами
- 25. В ходе дифференцировки клеток, продуцирующих антитела,
- 26. Так, при образовании полноразмерного готового к транскрипции
- 27. Аналогично перестраиваются гены для L-цепи иммуноглобулинов.
- 28. При антигенной стимуляции начинают размножаться и подвергаться
- 29. При этом частота спонтанных мутаций оказывается на
- 30. Таким образом, разнообразие антител достигается за счет
- 31. Сходный механизм соматической перегруппировки характерен и для
- 32. Этот белок имеет структурное сходство с иммуноглобулинами.
- 33. Подчеркнем еще раз, что перестройка генетического материала
- 34. Тяжелый комбинированный иммунодефицит
- 35. Тяжелый комбинированный иммунодефицит (ТКИД) — это
- 36. Начиная с младенческого возраста, эти больные подвержены
- 37. У больных наблюдается выраженная лимфопения, сопровождающаяся снижением
- 38. Продолжительность жизни больных при отсутствии лечения обычно
- 39. ТКИД делится на 2 группы: с сохранением
- 40. Наиболее частым генетическим типом ТКИД является
- 41. В шести интерлейкиновых рецепторных комплексах присутствует
- 42. У больных мальчиков с гемизиготными мутациями в
- 43. К такой же форме (T-, B+, НК-)
- 44. Форма (T-, B+, NK+) ТКИД также генетически
- 45. У 20-30% больных комбинированным иммунодефицитом присутствуют клетки-киллеры,
- 46. Более чем у половины таких больных обнаруживаются
- 47. Продукты этих генов являются активаторами рекомбиназы, необходимой
- 48. Многие ферменты репарации ДНК одновременно участвуют в
- 49. Для каждого из этих шагов необходимы продукты
- 50. Формы (T-, B-, NK+), сопровождающиеся повышенной чувствительностью
- 51. Продукты генов DCLRE1C и NHEJ участвуют в
- 52. (T-, B-, NK-)-форма комбинированного иммунодефицита, или наследственная
- 53. Это редкое аутосомно-рецессивное заболевание, встречающееся с частотой
- 54. АДА-недостаточность составляет около 15% всех случаев ТКИД
- 55. Наиболее часто болезнь проявляется уже в младенческом
- 56. При асимптомных формах АДА-недостаточности активность фермента в
- 57. Первое успешное лечение АДА-недостаточности методами генотерапии
- 58. Лечебный эффект длился несколько месяцев, после чего
- 59. В результате лечения состояние пациентки настолько улучшилось,
- 60. В настоящее время программа генотерапевтического лечения ADA-недостаточности
- 61. При этих условиях эффект от каждой процедуры
- 62. К числу тяжелых комбинированных иммунодефицитов относится синдром
- 63. При этом становится невозможным Т-зависимый иммунный ответ,
- 64. Первыми симптомами СГЛ являются упорная диарея, кандидоз
- 65. В настоящее время идентифицированы гены при двух
- 66. В настоящее время выделяют 5 групп комплементации
- 67. При редком СГЛ I типа на поверхности
- 68. Характерными клиническими проявлениями являются бронхоэктазы, эмфизема, панбронхиолиты, бронхиальная обструкция, носовые полипы и синуситы
- 69. СГЛ I типа также представляет собой гетерогенную
- 70. Продуктами этих генов являются АТФ-связывающие транспортные белки,
- 71. Простой вариабельный иммунодефицит
- 72. Простой вариабельный иммунодефицит (ПВИД) связан с
- 73. При этом у больных снижена секреция иммуноглобулинов
- 74. Клинически болезнь проявляется частыми бактериальными респираторными и
- 75. Это гетерогенная группа аутосомно-рецессивных заболеваний, обусловленных мутациями
- 76. Это гены поверхностного Т-клеточного рецептора, выполняющего
- 77. А также специфических поверхностных В-клеточных антигенов,
- 78. Хронический гранулематоз
- 79. Хронический гранулематоз (ХГ) обусловлен неспособностью нейтрофилов и
- 80. Главная причина дефектной функции фагоцитов заключается в
- 81. В основе патогенеза этого генетически гетерогенного заболевания
- 82. Эти дефекты приводят к снижению способности мононуклеарных
- 83. Заболевание обычно проявляется в первые два года
- 85. В последующем воспалительные гранулемы и абсцессы возникают
- 86. Появление гранулем связано с неспособностью фагоцитов к
- 87. Диагноз хронического гранулематоза подтверждается при выявлении низкой
- 88. Больные постоянно нуждаются в антибактериальной терапии, требующейся
- 89. либо (2) назначается чередование антибиотиков широкого спектра
- 90. В настоящее время идентифицированы 5 генов, мутации
- 91. Аутосомно-рецессивный цитохром b-негативный тип составляет не более
- 92. Остальные аутосомно-рецессивные типы заболевания являются цитохром
- 93. 90% аутосомных форм и около 30% всех
- 94. 97% мутантных аллелей в гене NCF1
- 95. Идентифицирован ген при клинически сходном синдроме
- 96. Инфантильный летальный агранулоцитоз , или болезнь
- 97. Клинические проявления характеризуются рецидивирующими бактериальными инфекциями
- 98. Вместе с тем, у больных отмечается резистентность
- 99. Синдромальные формы наследственного иммунодефицита
- 100. В некоторых случаях наследственный иммунодефицит может сочетаться с патологией других систем
- 101. Синдром рекуррентных инфекций, обусловленный гиперпродукцией IgE (гипер-IgE-синдром),
- 102. Больные имеют характерные грубые черты лица, аномальный
- 103. По аутосомно-рецессивному типу наследуются типы, обусловленные мутациями
- 104. Аутосомно-доминантный гипер-IgE-синдром, характеризующийся дополнительно комплексом специфических
- 105. При Х-сцепленном лимфопролиферативномй синдроме, или болезни Дункана,
- 106. У больных, перенёсших инфекционный мононуклеоз, нередко развиваются
- 107. В комплексный фенотип болезни Дункана входят тяжелый
- 108. Заболевание обусловлено мутациями в гене SH2D1A, кодирующем
- 109. Сигнальное SLAM-SLAM-связывание, возникающее при взаимодействии между
- 110. По крайней мере, 6 аллельных Х-сцепленных доминантных
- 111. Продукт гена IKBKG (IKK-гамма), относящийся к консервативному
- 112. Этот фактор присутствует во множестве клеток, в
- 113. Несвоевременная активация NFKB ассоциирована с
- 114. С другой стороны, полное или частичное ингибирование
- 115. Мутации в гене MYD88 являются причиной
- 116. Основной функцией адапторного белка MyD88 является индукция
- 117. Мутации в гене STK4 – стресс-индуцируемой
- 118. У больных наблюдается прогрессирующая потеря Т-клеток,
- 119. Stk4-киназа активируется в ответ на действие факторов
- 120. Не исключено, что эта форма синдромального иммунодефицита
- 121. К эпигенетическим болезням, по-видимому, может быть отнесен
- 122. Продуктом этого гена является метилтрансфераза 3В –
- 123. ICF-синдром характеризуется заметным иммунодефицитом, часто проявляющимся
- 124. Для большинства пациентов продолжительность жизни снижена из-за рецидивирующих бронхиальных и легочных инфекций
- 125. БЛАГОДАРЮ ЗА ВНИМАНИЕ
Слайд 1Наследственные иммунодефициты
В. Н. Горбунова
Санкт-Петербургский государственный педиатрический медицинский университет

Слайд 2Наследственные иммунодефицитные состояния — это гетерогенная группа болезней, характеризующаяся недостаточностью иммунной
системы вследствие генетических дефектов, приводящих к нарушениям процессов пролиферации, дифференцировки и функционирования клеток иммунокомпетентной системы

Слайд 3Клинические проявления иммунодефицитных состояний обусловлены снижением активности или неспособностью организма к
эффективному осуществлению реакций клеточного и/или гуморального звеньев иммунитета

Слайд 4Распространенность первичных наследственных иммунодефицитов среди населения существенно варьирует от 1 :
25000 до 1 : 50000.
Однако такие варианты врожденных иммунных дефектов,
как селективный дефицит IgA, встречаются с частотой 1:500 - 1:700

Слайд 5Недостаточность системы В-лимфоцитов и гуморального звена иммунитета выявляется у 50-75% из
общего числа больных с различными иммунодефицитными состояниями

Слайд 6В зависимости от преимущественного повреждения клеток иммунной системы все иммунодефициты разделены
на 4 группы

Слайд 7(1) Т-зависимые, клеточные с повреждением клеточного иммунитета, (2) В-зависимые, гуморальные с
повреждением гуморального иммунитета,
(3) А-зависимые с нарушениями системы фагоцитоза и
(4) комбинированные с поражением клеточного и гуморального звеньев иммунитета
 Т-зависимые, клеточные с повреждением клеточного иммунитета, (2) В-зависимые, гуморальные с повреждением гуморального иммунитета, (3)")
Слайд 8Важнейшая роль в производстве антител принадлежит генам иммуноглобулинов и Т-клеточных рецепторов,
синтезируемых соответственно В- и Т-лимфоцитами

Слайд 9В ходе созревания лимфоцитов от стволовой клетки до развития их специализированных
субпопуляций происходит уникальная перестройка этих генов.
Важнейшими регуляторами иммунных реакций являются цитокины (интерлейкины, интерфероны и др.), осуществляющие свое действие через специфические рецепторы

Слайд 10Цитокиновая система обеспечивает взаимодействие между лимфоцитами и фагоцитами, координируя тем самым
работу разнообразных клеток, задействованных в иммунных реакциях

Слайд 11 В основе развития наследственных иммунодефицитов могут лежать многие процессы, затрагивающие,
в частности, (1) созревание лимфоцитов, фагоцитов и других иммунных клеток,
(2) нарушения перестройки и экспрессии генов иммуноглобулинов и Т-клеточных рецепторов, а также
(3) недостаточность цитокиновой системы контроля иммунных реакций
 созревание")
Слайд 12При наиболее тяжелых формах комбинированного иммунодефицита нарушается процесс дифференцировки Т-лимфоцитов. Нарушения
дифференцировки В-лимфоцитов приводят к простому вариабельному иммунодефициту

Слайд 13При хроническом гранулематозе наблюдается недостаточность фагоцитарной системы иммунитета. Иммунодефицитные состояния входят
в состав нескольких десятков моногенных синдромов


Слайд 15Организм каждого человека может вырабатывать около 1011 различных антител, и очевидно,
что необходимая для этого информация значительно превышает размер генома

Слайд 16Разнообразие генов иммуноглобулинов, контролирующих синтез антител, достигается за счет их физической
перегруппировки в соматических клетках и повышенной частоты соматических мутаций

Слайд 17Изначально антитела кодируются относительно небольшим количеством генов, но в ходе созревания
В-клеток происходит уникальная соматическая перестройка с участием процессов рекомбинации и репарации ДНК

Слайд 18Эта перестройка сопровождается резким увеличением частоты соматических мутаций, что, в конечном
счете, и приводит к огромному разнообразию антител

Слайд 19Молекулы иммуноглобулинов формируются из двух тяжелых (H) и двух легких (L)
цепей, причем каждая из этих цепей состоит из двух сегментов – постоянного (С) и
вариабельного (V)
 и двух легких (L) цепей, причем каждая из")
Слайд 20Постоянный участок определяет класс иммуноглобулинов (M, G, A, E или D),
и его аминокислотная последовательность относительно стабильна среди иммуноглобулинов одного и того же класса
, и его аминокислотная последовательность")
Слайд 21Аминокислотная последовательность V-участков очень изменчива у разных антител. V-участки H- и
L-цепей определяют специфичность антитела, так как формируют сайт узнавания для соответствующего антигена

Слайд 22В половых клетках человека нет полноразмерных генов, кодирующих H- и L-цепи,
но существуют множества гомологичных генов, отстоящих друг от друга на сотни тысяч нуклеотидов

Слайд 23Так, локус для V-участка H-цепи состоит из трех сегментов V, D
и J.
В первом сегменте присутствует более 200 различных генов и, возможно, псевдогенов,
около 30 генов находятся в сегменте D и 9 – в сегменте J

Слайд 24Причем гены вариабельного участка перемежаются с генами константного участка иммуноглобулинов разных
типов.
Таким образом, общая площадь ДНК, занятая генами как H-, так и L-цепей иммуноглобулинов, составляет миллионы пар оснований

Слайд 25В ходе дифференцировки клеток, продуцирующих антитела, ДНК в локусах иммуноглобулинов перестраивается,
создавая функциональные гены
для H- и L-цепей

Слайд 26Так, при образовании полноразмерного готового к транскрипции гена для H-цепи происходит
разрыв двойной спирали ДНК с последующим соединением концов таким образом, что различные варианты генов из V-, D- и J-сегментов объединяются, при этом промежуточные участки ДНК удаляются

Слайд 27Аналогично перестраиваются гены для L-цепи иммуноглобулинов. При неточном соединении сегментов генов
в процессе соматической перестройки могут возникать делеции или инсерции
(N-последовательности), обеспечивающие дополнительное разнообразие антител

Слайд 28При антигенной стимуляции начинают размножаться и подвергаться частым точечным мутациям в
пределах перестроенной последовательности ДНК те
В-лимфоциты, которые производят антитела, имеющие сродство с данным антигеном

Слайд 29При этом частота спонтанных мутаций оказывается на 2-3 порядка выше, чем
где-либо в геноме.
Эти мутации, меняя аминокислотную последовательность в вариабельном участке антитела, обеспечивают «тонкую настройку» его сродства к антигену

Слайд 30Таким образом, разнообразие антител достигается за счет (1) использования двух различных
цепей (H и L) иммуноглобулинов, перегруппированных за счет присоединения различных вариабельных сегментов (V, D и J),
(2) неточного соединения этих сегментов и (3) соматических мутаций вариабельного участка
 использования двух различных цепей (H и L)")
Слайд 31Сходный механизм соматической перегруппировки характерен и для семейства генов Т-клеточного рецептора
(TCR), продуктом которых является трансмембранный гликопротеин, играющий ключевую роль в узнавании антигенов и функционировании Т-лимфоцитов
, продуктом которых является")
Слайд 32Этот белок имеет структурное сходство с иммуноглобулинами. Все его цепи имеют
как постоянные, так и вариабельные участки, причем в образовании последних также участвуют V-, D- и J-сегменты.
Однако в отличие от иммуноглобулинов частота соматических мутаций в перестраиваемых вариабельных участках гена TCR сохраняется в пределах нормы

Слайд 33Подчеркнем еще раз, что перестройка генетического материала происходит только в В-
и Т-клетках
и касается исключительно генов иммуноглобулинов и Т-клеточных рецепторов.
Остальная часть генома в ходе развития и дифференцировки остается стабильной


Слайд 35Тяжелый комбинированный иммунодефицит (ТКИД) — это гетерогенная группа болезней, обусловленных нарушениями
клеточного и гуморального иммунитета
 — это гетерогенная группа болезней, обусловленных нарушениями клеточного и гуморального иммунитета")
Слайд 36Начиная с младенческого возраста, эти больные подвержены рекуррентным инфекционным процессам, развивающимся
вследствие активации
условно-патогенных микроорганизмов, включая Candida albicans, Pneumocystis carinii, цитомегаловирусы и многие другие

Слайд 37У больных наблюдается выраженная лимфопения, сопровождающаяся снижением или полным отсутствием иммуноглобулинов.
Общей характеристикой всех типов комбинированного иммунодефицита является нарушение дифференцировки Т-лимфоцитов и отсутствие опосредуемого ими клеточного иммунитета

Слайд 38Продолжительность жизни больных при отсутствии лечения обычно не превышает одного года.
Чаще всего, единственная возможность терапии этих заболеваний — трансплантация костного мозга.
Общая частота всех типов ТКИД составляет 1:75000 новорожденных

Слайд 39ТКИД делится на 2 группы: с сохранением и без сохранения В-лимфоцитов
– B+ТКИД и B-ТКИД.
В каждой из этих двух групп выделяют формы, при которых могут присутствовать или отсутствовать натуральные клетки-киллеры (НК), относящиеся к системе врожденного иммунитета и не требующие, в отличие от Т-киллеров, каскада реакций антигенной презентации

Слайд 40Наиболее частым генетическим типом ТКИД является Х-сцепленный комбинированный иммунодефицит, обусловленный
мутациями в гене гамма-рецептора интерлейкина 2 — IL2RG.
Этот тип относится к первой группе комбинированных иммунодефицитов, так как у больных мальчиков отсутствуют Т-клетки и НК, но присутствуют В-клетки (T-, B+, НК-)

Слайд 41В шести интерлейкиновых рецепторных комплексах присутствует общая гамма-цепь, кодируемая геном
IL2RG.
Все они являются гетеродимерами и взаимодействует с различными интерлейкин-специфическими альфа-цепями

Слайд 42У больных мальчиков с гемизиготными мутациями в гене IL2RG функция этих
комплексов частично или полностью блокирована, что приводит к тяжелым нарушениям регуляции гомеостаза иммунной системы со стороны цитокиновой системы

Слайд 43К такой же форме (T-, B+, НК-) относится аутосомно-рецессивный B+ТКИД, обусловленный
мутациями в гене JAK3.
Продукт этого гена относится к семейству тирозинкиназ, участвующих в опосредуемой цитокиновыми рецепторами внутриклеточной сигнальной трансдукции
 относится аутосомно-рецессивный B+ТКИД, обусловленный мутациями в гене JAK3.")
Слайд 44Форма (T-, B+, NK+) ТКИД также генетически гетерогенна и может быть
вызвана дефектами цитокиновых рецепторов (IL7R), негативных регуляторов
сигнальных путей, опосредуемых цитокиновыми рецепторами (CD45)
или белков-регуляторов дифференцировки Т-клеток (CD3D)
 ТКИД также генетически гетерогенна и может быть вызвана дефектами цитокиновых рецепторов")
Слайд 45У 20-30% больных комбинированным иммунодефицитом присутствуют клетки-киллеры, при этом отсутствуют Т-
и В-лимфоциты – форма (T-, B-, НК+)

Слайд 46Более чем у половины таких больных обнаруживаются мутации в одном из
двух соседних синхронно регулируемых генов
RAG1 и RAG2,
расположенных в области 11p13

Слайд 47Продукты этих генов являются активаторами рекомбиназы, необходимой для перестройки V-, D-
и J-сегментов, которая происходит при образовании функциональных генов для
H- и L-цепей иммуноглобулинов (так называемая
V(D)J-рекомбинация)

Слайд 48Многие ферменты репарации ДНК одновременно участвуют в V(D)J-рекомбинации. Этот процесс начинается
с образования двунитевого разрыва, который, в свою очередь, состоит из двух шагов — образования однонитевого разрыва и шпилечной структуры ДНК
J-рекомбинации. Этот процесс начинается с образования двунитевого разрыва,")
Слайд 49Для каждого из этих шагов необходимы продукты генов RAG1 и RAG2.
Различные мутации в этих генах могут приводить к разным аллельным иммунодефицитным состояниям таким, например, как синдром Оменна

Слайд 50Формы (T-, B-, NK+), сопровождающиеся повышенной чувствительностью к ионизирующему облучению, могут
быть обусловлены мутациями в одном из двух генов
DCLRE1C или NHEJ1.
В последнем случае у больных отмечается микроцефалия и задержка роста
, сопровождающиеся повышенной чувствительностью к ионизирующему облучению, могут быть обусловлены мутациями в")
Слайд 51Продукты генов DCLRE1C и NHEJ участвуют в репарации ДНК и V(D)J-рекомбинации
на стадиях образования шпилечной структуры ДНК и негомологичного соединения концов соответственно
J-рекомбинации на стадиях образования шпилечной")
Слайд 52(T-, B-, NK-)-форма комбинированного иммунодефицита, или наследственная недостаточность аденозиндезаминазы (АДА) клинически
является наиболее тяжелой и сопровождается нарушением функционирования тимуса
-форма комбинированного иммунодефицита, или наследственная недостаточность аденозиндезаминазы (АДА) клинически является наиболее тяжелой и")
Слайд 53Это редкое аутосомно-рецессивное заболевание, встречающееся с частотой 1:100 000. АДА необходима
для расщепления пуринов.
При ее отсутствии в крови больных накапливается промежуточный метаболит – 2-дезоксиаденозин, который препятствует нормальному созреванию и пролиферации
T- и B-лимфоцитов

Слайд 54АДА-недостаточность составляет около 15% всех случаев ТКИД и около трети –
его аутосомно-рецессивных форм. Клинический полиморфизм заболевания определяется остаточной активностью фермента

Слайд 55Наиболее часто болезнь проявляется уже в младенческом возрасте и может быстро
приводить к летальному исходу.
У 10-15% больных первые признаки заболевания появляются во втором полугодии жизни.
Значительно реже отмечается более позднее начало появления первых симптомов – в детском или даже взрослом возрасте

Слайд 56При асимптомных формах АДА-недостаточности активность фермента в ядерных клетках крови может
составлять от 5 до 80% по сравнению с нормой, а в эритроцитах – практически отсутствует. При своевременной постановке диагноза АДА-недостаточность успешно лечится с использованием методов генной терапии

Слайд 57Первое успешное лечение АДА-недостаточности методами генотерапии было осуществлено более 20 лет
назад в США.
Девочке 4-х лет, страдающей этим редким заболеванием, были пересажены ее собственные лимфоциты, в которые предварительно в условиях культивирования был введен нормальный ген ADA

Слайд 58Лечебный эффект длился несколько месяцев, после чего процедуру повторяли с интервалом
в 3-5 месяцев.
На протяжении трех лет терапии в общей сложности было проведено 23 внутривенных трансфузии ADA-трансформированных Т-лимфоцитов без видимых неблагоприятных эффектов

Слайд 59В результате лечения состояние пациентки настолько улучшилось, что она смогла вести
нормальный образ жизни.
Столь же успешным оказалось лечение и других подобных пациентов, проводимое в США, Италии, Франции, Великобритании и Японии

Слайд 60В настоящее время программа генотерапевтического лечения ADA-недостаточности модифицирована таким образом, что
генетическая конструкция, содержащая нормальный ген ADA, вводится не в зрелые T-лимфоциты, а в стволовые клетки костного мозга

Слайд 61При этих условиях эффект от каждой процедуры реинфузии пролонгируется. После 2-3
лет подобной терапии пациенты, как правило, уже не нуждаются в повторных введениях модифицированных клеток

Слайд 62К числу тяжелых комбинированных иммунодефицитов относится синдром «голых» лимфоцитов (СГЛ), при
котором на поверхности антигенпрезентирующих клеток лимфоцитов не экспрессируются молекулы главного комплекса гистосовместимости – HLA
, при котором на поверхности антигенпрезентирующих")
Слайд 63При этом становится невозможным Т-зависимый иммунный ответ, хотя количество Т- и
В-лимфоцитов в крови нормальное.
Заболевание проявляется в возрасте 3—6 месяцев различными инфекциями и задержкой физического развития

Слайд 64Первыми симптомами СГЛ являются упорная диарея, кандидоз кожи и слизистых, интерстициальная
пневмония и различные бактериальные инфекции

Слайд 65В настоящее время идентифицированы гены при двух типах СГЛ – I
и II.
Более распространенным является II тип СГЛ – гетерогенная группа редких аутосомно-рецессивных комбинированных иммунодефицитов, обусловленных мутациями в трансактивирующих регуляторных генах, экспрессия которых необходима для транскрипции генов класса II HLA

Слайд 66В настоящее время выделяют 5 групп комплементации СГЛ II типа, обусловленных
мутациями в генах различных регуляторных факторов, трансактиваторов и взаимодействующих с ними белков
(MHC2TA – группа A;
RFXANK – группа B;
RFX5 – группы C и E;
RFXAP – группа D)

Слайд 67При редком СГЛ I типа на поверхности лимфоцитов отсутствуют молекулы класса
I HLA, в то время как молекулы класса II присутствуют.
В этом случае хронические бактериальные инфекции, начинающиеся, как правило, в первом десятилетии жизни ограничиваются верхними и нижними дыхательными путями

Слайд 68Характерными клиническими проявлениями являются бронхоэктазы, эмфизема, панбронхиолиты, бронхиальная обструкция, носовые полипы
и синуситы

Слайд 69СГЛ I типа также представляет собой гетерогенную группу заболеваний, обусловленных мутациями
в трех тандемно расположенных в области 6p21.32 генах –
TAP1, TAP2 и TAPAB

Слайд 70Продуктами этих генов являются АТФ-связывающие транспортные белки, участвующие в транслокации необходимых
цитоплазматических пептидов к ожидающим их в эндоплазматическом ретикулуме молекулам HLA класса I


Слайд 72Простой вариабельный иммунодефицит (ПВИД) связан с нарушением передачи сигналов от Т-
к В-клеткам. Ведущим проявлением заболевания является гипогаммаглобулинемия
 связан с нарушением передачи сигналов от Т- к В-клеткам. Ведущим проявлением")
Слайд 73При этом у больных снижена секреция иммуноглобулинов IgG, IgA и в
половине случаев – IgM, а также наблюдаются нарушения дифференцировки В-лимфоцитов, хотя содержание В-клеток может быть в пределах нормы.
У некоторых больных отмечается частичная Т-клеточная дисфункция

Слайд 74Клинически болезнь проявляется частыми бактериальными респираторными и кишечными инфекциями, которые могут
развиваться в любом возрасте
(с пиками встречаемости в первом и третьем десятилетиях жизни).
От 10 до 20% больных имеют семейную историю заболевания

Слайд 75Это гетерогенная группа аутосомно-рецессивных заболеваний, обусловленных мутациями в генах сигнальных молекул,
локализованных на поверхности Т- или В-лимфоцитов.
В настоящее время идентифицированы гены, мутации в которых приводят к 8 различным генетическим типам этого заболевания

Слайд 76Это гены поверхностного Т-клеточного рецептора, выполняющего функции индуцибельного ко-стимулятора Т-клеток (ICOS);
специфических
В-клеточных рецепторов,
участвующих в активации В-лимфоцитов (TNFRSF13B и TNFRSF13C);
; специфических В-клеточных рецепторов, участвующих")
Слайд 77А также специфических поверхностных В-клеточных антигенов, входящих в состав В-клеточного ко-рецепторного
комплекса, главной функцией которого является понижение порога активации
В-клеточного антигенного рецептора
(CD19; CD20, CD21, CD8), и некоторых других


Слайд 79Хронический гранулематоз (ХГ) обусловлен неспособностью нейтрофилов и моноцитов лизировать поглощённые микроорганизмы
с каталазоположительными свойствами,
т.е. вырабатывающие перекись водорода (золотистый стафилококк, серрации, аспергилус, кандиды),
а также большинство грамотрицательных бактерий
 обусловлен неспособностью нейтрофилов и моноцитов лизировать поглощённые микроорганизмы с каталазоположительными свойствами, т.е.")
Слайд 80Главная причина дефектной функции фагоцитов заключается в нарушении окислительного метаболизма в
связи с наличием блока в процессах образования супероксиданиона и
перекиси водорода

Слайд 81В основе патогенеза этого генетически гетерогенного заболевания лежат дефекты структурных или
регуляторных субъединиц
белков фагоцитарного НАДФ-оксидазного комплекса (phox), участвующих в образовании бактерицидной перфорации, отсутствие или дефектный
цитохром b558

Слайд 82Эти дефекты приводят к снижению способности мононуклеарных клеток выступать в качестве
антигенпрезентирующих единиц за счет нарушения процессинга и презентации антигена

Слайд 83Заболевание обычно проявляется в первые два года жизни ребенка и у
девочек протекает легче.
Характерным является развитие гнойничковых инфильтратов
в коже и
экзематозного дерматита вокруг рта, носа и ушей

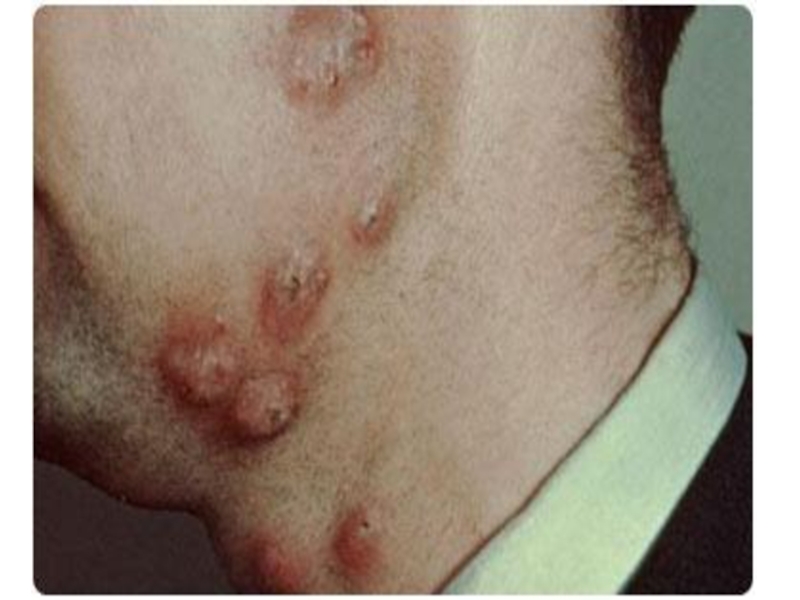
Слайд 85В последующем воспалительные гранулемы и абсцессы возникают в различных органах (чаще
всего в легких), при этом выявляются генерализованная лимфаденопатия, гепато- и спленомегалия
, при")
Слайд 86Появление гранулем связано с неспособностью фагоцитов к разрушению и перевариванию поглощенных
микроорганизмов. Частыми инфекционными осложнениями являются грибковые поражения легких, внутренних органов, кожи, слизистых

Слайд 87Диагноз хронического гранулематоза подтверждается при выявлении низкой активности ферментов гексозомонофосфатного шунта
и дефекта продукции перекисных соединений нейтрофилами
(с помощью теста с нитросиним тетразолием)

Слайд 88Больные постоянно нуждаются в антибактериальной терапии, требующейся даже в периоде ремиссии.
В зависимости от степени восприимчивости к инфекциям и чувствительности инфекционного агента к препаратам, больные получают постоянно либо
(1) триметоприм – сульфаметоксазол,

Слайд 89либо (2) назначается чередование антибиотиков широкого спектра действия (цефалоспоринов, полусинтетических пенициллинов,
оксихинолонов и др.)
в сочетании с антимикотическими препаратами.
При абсцессах легких и других внутренних органов показана интенсивная длительная антибактериальная терапия одновременно несколькими препаратами
 назначается чередование антибиотиков широкого спектра действия (цефалоспоринов, полусинтетических пенициллинов, оксихинолонов и др.) в")
Слайд 90В настоящее время идентифицированы 5 генов, мутации в которых приводят к
хроническому гранулематозу.
Самым частым является
Х-сцепленный тип, объясняющий около 60% всех случаев заболевания.
При этом цитохром b-негативном типе мутантным является ген CYBB, кодирующий белок p91-phox

Слайд 91Аутосомно-рецессивный цитохром b-негативный тип составляет не более 3% случаев заболевания. Он
обусловлен мутациями в гене CYBA,
кодирующим белок p22-phox

Слайд 92Остальные аутосомно-рецессивные типы заболевания являются цитохром b-позитивными и могут быть обусловлены
мутациями в генах нейтрофильных цитоплазматических факторов фагоцитарной оксидазы

Слайд 9390% аутосомных форм и около 30% всех случаев заболевания обусловлены мутациями
в гене нейтрофильного цитоплазматического фактора
p47-phox (NCF1),
в остальных случаях –
p67-phox (NCF2) и
p40-phox (NCF4)

Слайд 9497% мутантных аллелей в гене NCF1 представлены одной делецией 2
нуклеотидов – 75GT. Присутствие мажорной мутации в гене NCF1 существенно облегчает молекулярную диагностику данного типа заболевания

Слайд 95Идентифицирован ген при клинически сходном синдроме нейтрофильного иммунодефицита – RAC2. Его
продуктом также является один из белков фагоцитарного NADPH-оксидазного комплекса, взаимодействующий с p67-phox

Слайд 96Инфантильный летальный агранулоцитоз , или болезнь Костмана – заболевание с аутосомно-рецессивным
типом наследования, обусловленное мутациями в гене HAX1, продукт которого, предположительно, вовлечен в ингибирование апоптоза.
В костном мозге больных отсутствуют клетки нейтрофильного ряда более зрелые, чем промиелоциты

Слайд 97 Клинические проявления характеризуются рецидивирующими бактериальными инфекциями кожи в области волосистой
части головы, перианальной области, слизистых оболочек, придаточных пазух носа, лёгких, а также развитием сепсиса и, нередко, остеомиелита

Слайд 98Вместе с тем, у больных отмечается резистентность к вирусным и грибковым
инфекциям.
При лабораторном исследовании количество нейтрофилов в крови менее 300 в 1 мкл при нормальном содержании лейкоцитов и резком моноцитозе с эозинофилией.
Прогноз для жизни неблагоприятный. Больные часто погибают от сепсиса в первые 3 года жизни


Слайд 100В некоторых случаях наследственный иммунодефицит может сочетаться с патологией других систем

Слайд 101Синдром рекуррентных инфекций, обусловленный гиперпродукцией IgE (гипер-IgE-синдром), или синдром Джоба является
первичным иммунодефицитом, проявляющимся хронической экземой, рекуррентными стафилококковыми инфекциями, высоким содержанием сывороточного IgE, лейкоцитозом и эозинофилией
, или синдром Джоба является первичным иммунодефицитом, проявляющимся хронической")
Слайд 102Больные имеют характерные грубые черты лица, аномальный прикус в сочетании с
гиперрастяжимостью кожи.
Этот синдром также представляет собой гетерогенную группу наследственных заболеваний

Слайд 103По аутосомно-рецессивному типу наследуются типы, обусловленные мутациями в двух генах –
DOCK8 и CD40.
Ген DOCK8 активно экспрессируется в моноцитах, а также в
Т- и В-лимфоцитах.
Продуктом гена CD40 является поверхностный антиген, присутствующий во всех зрелых
В-клетках и моноцитах

Слайд 104Аутосомно-доминантный гипер-IgE-синдром, характеризующийся дополнительно комплексом специфических соединительнотканных и скелетных нарушений, обусловлен
мутациями в гене STAT3.
Продуктом этого гена является транскрипционный фактор, активирующийся под действием некоторых интерлейкинов и
факторов роста

Слайд 105При Х-сцепленном лимфопролиферативномй синдроме, или болезни Дункана, наблюдаются тяжелые нарушения регуляции
иммунной системы, часто возникающие после инфицирования, например, вирусом Эпштейна-Барр

Слайд 106У больных, перенёсших инфекционный мононуклеоз, нередко развиваются длительное лихорадочное состояние, лимфаденопатия,
лимфоцитоз в периферической крови, гепато- и спленомегалия, гепатит.
Позднее формируется
В-клеточная лимфома,
чаще в терминальных отделах тонкой кишки, которая и может служить причиной летального исхода

Слайд 107В комплексный фенотип болезни Дункана входят тяжелый или фатальный мононуклеоз, приобретенная
гипогаммаглобулинемия, гемофагоцитарный лимфогистоцитоз, апластическая анемия, лимфоматоидный гранулематоз, злокачественная лимфома

Слайд 108Заболевание обусловлено мутациями в гене SH2D1A, кодирующем белок сигнальной активации лимфоцитов
(SLAM), участвующий в бинаправленной стимуляции Т- и В-клеток.
В активированном состоянии он опосредует экспансию
Т-клеток во время иммунного ответа, индуцирует продукцию гамма-интерферона и изменяет функциональный профиль Т-клеток
, участвующий в бинаправленной")
Слайд 109Сигнальное SLAM-SLAM-связывание, возникающее при взаимодействии между В-клетками и между В- и
Т-клетками, ускоряет экспансию и дифференцировку В-лимфоцитов

Слайд 110По крайней мере, 6 аллельных Х-сцепленных доминантных иммунодефицитов, которые могут сочетаться
с эктодермальной дисплазией, обусловлены мутациями в гене IKBKG. Гемизиготные мутации у мужчин приводят к эмбриональной летальности, поэтому болеют только гетерозиготные женщины

Слайд 111Продукт гена IKBKG (IKK-гамма), относящийся к консервативному семейству NEMO-киназ, входит в
состав киназного комплекса, принимающего участие в разрушении транскрипционного ядерного фактора NFKB
, относящийся к консервативному семейству NEMO-киназ, входит в состав киназного комплекса, принимающего")
Слайд 112Этот фактор присутствует во множестве клеток, в которых экспрессируются цитокины, хемокины,
факторы роста, молекулы клеточной адгезии и некоторые белки острой фазы.
NFKB активируется под действием многих стимулов, включая цитокины, свободные радикалы, УФ-облучение, бактериальные или вирусные продуценты

Слайд 113 Несвоевременная активация NFKB ассоциирована с воспалительными процессами при аутоиммунных артритах,
бронхиальной астме, септическом шоке, фиброзе легких, гломерулонефрите, атеросклерозе, СПИДе

Слайд 114С другой стороны, полное или частичное ингибирование NFKB связано с процессами
апоптоза, нарушением дифференцировки иммунных клеток и задержкой клеточного роста

Слайд 115Мутации в гене MYD88 являются причиной развития аутосомно-рецессивного синдрома рекуррентных бактериальных
гнойных инфекций,
включая пневмококковые инвазии

Слайд 116Основной функцией адапторного белка MyD88 является индукция активации транскрипционного фактора NFKB.
В гене MYD88 идентифицированы две мажорные мутации —
E52del и R196C

Слайд 117Мутации в гене STK4 – стресс-индуцируемой серин/треонинкиназы – ответственны за развитие
аутосомно-рецессивного синдрома
Т-клеточного иммунодефицита, сочетающегося с врожденными пороками сердца

Слайд 118У больных наблюдается прогрессирующая потеря Т-клеток, рекуррентные бактериальные, вирусные и грибковые
инфекции, бородавки, абсцессы и аутоиммунные проявления в сочетании с дефектами межпредсердной перегородки

Слайд 119Stk4-киназа активируется в ответ на действие факторов роста, многих химических соединений,
теплового шока, апоптоз-индуцирующих агентов.
Более того, эта киназа осуществляет апоптоз-зависимое фосфорилирование гистонов,
что указывает на возможность ее участия в конденсации хроматина

Слайд 120Не исключено, что эта форма синдромального иммунодефицита обусловлена нарушением регуляции эпигенетических
процессов в ДНК, опосредуемых химическими модификациями гистонов

Слайд 121К эпигенетическим болезням, по-видимому, может быть отнесен аутосомно-рецессивный синдром иммунной недостаточности-центромерной
нестабильности-лицевых аномалий – ICF-синдром, так как он обусловлен мутациями в гене DNMT3B

Слайд 122Продуктом этого гена является метилтрансфераза 3В – фермент, принимающий участие в
одной из главных эпигенетических модификаций генома —
метилировании de novo

Слайд 123ICF-синдром характеризуется заметным иммунодефицитом, часто проявляющимся в виде хронических респираторных и
желудочно-кишечных инфекций,
в сочетании с характерными лицевыми аномалиями, включающими гипертелоризм, эпикант и низкую посадку ушей

Слайд 124Для большинства пациентов продолжительность жизни снижена из-за рецидивирующих бронхиальных и легочных
инфекций







