- Главная
- Разное
- Дизайн
- Бизнес и предпринимательство
- Аналитика
- Образование
- Развлечения
- Красота и здоровье
- Финансы
- Государство
- Путешествия
- Спорт
- Недвижимость
- Армия
- Графика
- Культурология
- Еда и кулинария
- Лингвистика
- Английский язык
- Астрономия
- Алгебра
- Биология
- География
- Детские презентации
- Информатика
- История
- Литература
- Маркетинг
- Математика
- Медицина
- Менеджмент
- Музыка
- МХК
- Немецкий язык
- ОБЖ
- Обществознание
- Окружающий мир
- Педагогика
- Русский язык
- Технология
- Физика
- Философия
- Химия
- Шаблоны, картинки для презентаций
- Экология
- Экономика
- Юриспруденция
Митохондриальные энцефалопатии презентация
Содержание
- 1. Митохондриальные энцефалопатии
- 2. Mitochondrial disease may cause any symptom in
- 3. Митохондрия является органеллой, которая присутствует практически в
- 5. Нарушения клеточной энергетики приводят к полисистемным заболеваниям.
- 6. Распространенность митохондриальных заболеваний составляет 11.5/100000. Среди
- 8. Дыхательная цепь локализуется на внутренней мембране
- 9. В клетке находятся от нескольких сотен до
- 10. На основе этиологии и патогенеза существует классификация
- 11. ТИП НАСЛЕДОВАНИЯ Митохондриальная генетика отличается от менделевской
- 12. Основные особенности митохондриальных цитопатий Выраженный полиморфизм клинических
- 13. КРИТЕРИИ ДИАГНОСТИКИ МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ Клинические : миопатический
- 14. Признаки поражения соединительной ткани (гипермобильный синдром, гиперэластичность
- 15. Биохимические тесты лактат-пируват-ацидоз . НОРМАЛЬНЫЕ ПОКАЗАТЕЛИ
- 16. НАИБОЛЕЕ ЧАСТО ВСТРЕЧАЮЩИЕСЯ В ДЕТСКОМ ВОЗРАСТЕ КЛИНИЧЕСКИЕ
- 17. Синдром Лея Подострая некротизирующая энцефаломиелопатия – описана
- 18. Болезнь Лея (подострая некротизирующая энцефаломиелопатия) Мутации митохондриальных или ядерных генов, отвечающие за возникновение болезни Лея.
- 19. Синдром MERRF Мультисистемное заболевание – основные
- 20. Синдром Кернса-Сейра Описан в 1958 году у
- 21. У большинства пациентов встречаются -низкий рост -потеря
- 22. МИТОХОНДРИАЛЬНАЯ ЭНЦЕФАЛОМИОПАТИЯ, ЛАКТАТ-АЦИДОЗ И ИНСУЛЬТОПОДОБНЫЕ ПАРОКСИЗМЫ (MELAS)
- 24. ОСОБЕННОСТИ ИНСУЛЬТА ПРИ СИНДРОМЕ MELAS Частая локализация
- 25. Количественный анализ церебрального кровотока, исследованный с помощью
- 27. Т.О. суммируя все вышеизложенное, нейрорадиологические исследования
- 28. ЛЕЧЕНИЕ МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ Нет достоверных подтверждений, что
- 29. ИСПОЛЬЗУЕМЫЕ ПРЕПАРТЫ КоэнзимQ10 – от 90
- 30. Кортикостероиды, минералокортикоиды - при развитии надпочечниковой недостаточности
- 31. Имеется большой пул заболеваний, причиной которых является
- 32. Важность своевременной диагностики митохондриальных болезней, поиска клинических
- 33. СПАСИБО ЗА ВНИМАНИЕ!!!

Слайд 2Mitochondrial disease may cause any symptom in any tissue at any
age by any inheritance. A Munnich.
Митохондриальные болезни могут быть причиной любого симптома в любой ткани в любом возрасте с любым типом наследования.
Митохондриальные болезни могут быть причиной любого симптома в любой ткани в любом возрасте с любым типом наследования.

Слайд 3Митохондрия является органеллой, которая присутствует практически в каждой клетке, за исключением
зрелых эритроцитов. Поэтому митохондриальные болезни могут поражать любые системы органов.
В связи с этим правильнее называть эти состояния митохондриальными цитопатиями [Sarnat HB,Menkes JH 2006]
Митохондриальные нарушения – это обширная группа патологических состояний, обусловленных генетическими, структурными, биохимическими дефектами митохондрий, нарушением тканевого дыхания и, как следствие, недостаточностью энергетического обмена. Митохондриальные дыхательные цепи- главный конечный путь аэробного метаболизма.
В связи с этим правильнее называть эти состояния митохондриальными цитопатиями [Sarnat HB,Menkes JH 2006]
Митохондриальные нарушения – это обширная группа патологических состояний, обусловленных генетическими, структурными, биохимическими дефектами митохондрий, нарушением тканевого дыхания и, как следствие, недостаточностью энергетического обмена. Митохондриальные дыхательные цепи- главный конечный путь аэробного метаболизма.

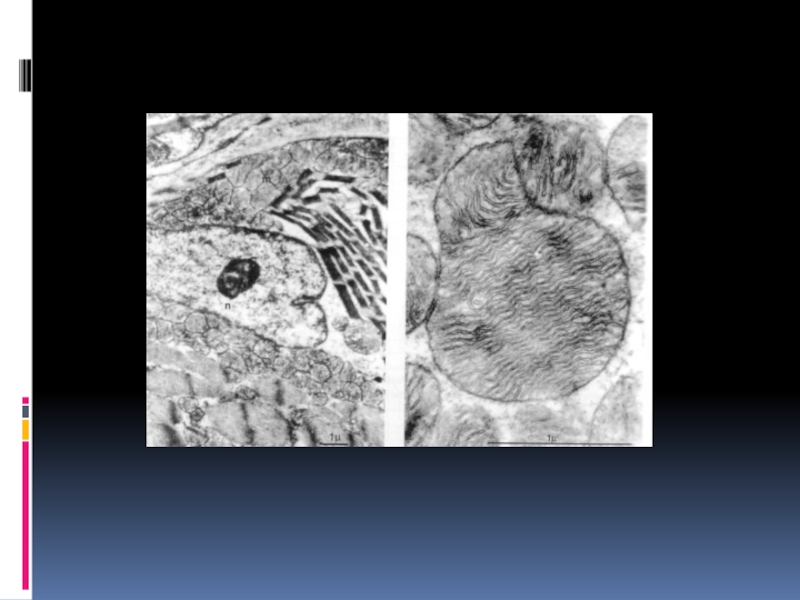
Слайд 5Нарушения клеточной энергетики приводят к полисистемным заболеваниям. В первую очередь страдают
органы и ткани, наиболее энергозависимые – нервная система (энцефалопатии, полиневропатии), мышечная система (миопатии), сердце (кардиомиопатии), почки,печень,эндокринная система и другие. До самого недавнего времени все эти заболевания определялись под многочисленными масками других патологических форм.
К настоящему времени выявлено более 200 заболеваний, причиной которых являются мутации митохондриальной ДНК. Митохондриальные болезни могут быть обусловлены патологией как митохондриального, так и ядерного генома.
К настоящему времени выявлено более 200 заболеваний, причиной которых являются мутации митохондриальной ДНК. Митохондриальные болезни могут быть обусловлены патологией как митохондриального, так и ядерного генома.

Слайд 6
Распространенность митохондриальных заболеваний составляет 11.5/100000. Среди жителей Испании в возрасте свыше
14 лет – 5.7/100000 [ Arpa et al, 2003]
Мутации мтДНК были выявлены у
1/8 000 населения [Chinnery PF, DiMauro S et all 2004]
Мутации мтДНК были выявлены у
1/8 000 населения [Chinnery PF, DiMauro S et all 2004]

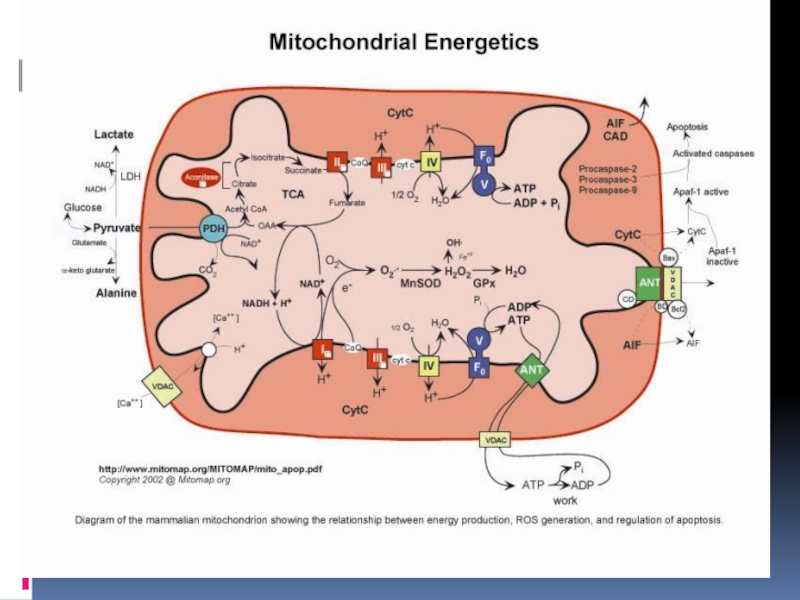
Слайд 8
Дыхательная цепь локализуется на внутренней мембране митохондрий и включает в себя
пять мультиферментных комплекса, каждый из которых в свою очередь состоит из нескольких десятков субъединиц. Митохондриальная ДНК кодирует только 13 из белковых субъединиц дыхательной цепи, 2 белковых субъединицы мРНК и 22 митохондриальных тРНК. Ядерный геном кодирует более 90% митохондриальных белков. Конечным результатом окислительного фосфорилирования, происходящего в комплексах 1-Y является производство энергии. (АТФ).

Слайд 9В клетке находятся от нескольких сотен до тысяч органелл – митохондрий,
содержащих от 2 до 10 кольцевых молекул митохондриальной ДНК,
способных к репликации, транскрипции и трансляции,
независимо от ядерной ДНК.

Слайд 10На основе этиологии и патогенеза существует классификация митохондриальных болезней
1.
Болезни, обусловленные дефектами мтДНК:
а) точковыми мутациями;
б)делециями;
в)изолированными дупликациями или в сочетании с делециями;
2. Болезни, обусловленные дефектами ядерной ДНК:
а) мутациями, нарушающими работу электронтранспортной цепи митохондрий;
б) мутациями, нарушающими окислительное фосфорилирование
в) мутациями, вызывающими дефекты ферментов цикла Кребса
г) мутациями, нарушающими утилизацию субстратов
д) мутациями, нарушающими транспорт субстратов
3. Болезни, обусловленные дефектами мтДНК, которые вызваны нарушением ядерной ДНК:
а) тканеспецифическими делециями или дупликациями мтДНК;
б) истощением (делецией) мтДНК.
а) точковыми мутациями;
б)делециями;
в)изолированными дупликациями или в сочетании с делециями;
2. Болезни, обусловленные дефектами ядерной ДНК:
а) мутациями, нарушающими работу электронтранспортной цепи митохондрий;
б) мутациями, нарушающими окислительное фосфорилирование
в) мутациями, вызывающими дефекты ферментов цикла Кребса
г) мутациями, нарушающими утилизацию субстратов
д) мутациями, нарушающими транспорт субстратов
3. Болезни, обусловленные дефектами мтДНК, которые вызваны нарушением ядерной ДНК:
а) тканеспецифическими делециями или дупликациями мтДНК;
б) истощением (делецией) мтДНК.

Слайд 11ТИП НАСЛЕДОВАНИЯ
Митохондриальная генетика отличается от менделевской в трех важнейших аспектах:
Материнское наследование
- всю цитоплазму, вместе с находящимися в ней органеллами потомки получают вместе с яйцеклеткой.
Гетероплазмия- одновременное существование в клетке нормального (дикий) и мутантного типов ДНК
Митотическая сегрегация- оба типа мтДНК в процессе деления клетки могут распределяться случайным образом между дочерними клетками.
Гетероплазмия- одновременное существование в клетке нормального (дикий) и мутантного типов ДНК
Митотическая сегрегация- оба типа мтДНК в процессе деления клетки могут распределяться случайным образом между дочерними клетками.

Слайд 12Основные особенности митохондриальных цитопатий
Выраженный полиморфизм клинических симптомов
Мультисистемный характер поражения
Вариабельность течения
Прогрессирование
Неадекватное реагирование
на применяемую терапию

Слайд 13КРИТЕРИИ ДИАГНОСТИКИ МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ
Клинические :
миопатический симптомокомплекс (непереносимость физических нагрузок, мышечная слабость,
снижение мышечного тонуса),
судороги (миоклонические или мультифокальные судорог),
мозжечковый синдром (атаксия, интенционный тремор),
поражение глазодвигательных нервов (птоз, наружная офтальмоплегия),
полиневропатия,
инсультоподобные пароксизмы, мигренеподобные головные боли,
черепно-лицевая дисморфия,
дисметаболические проявления (рвота,эпизоды летаргии,комы),
дыхательные нарушения (апноэ, гипервентилляция, тахипноэ)
поражение сердца, печени, почек,
прогрессирующее течение заболевания.
судороги (миоклонические или мультифокальные судорог),
мозжечковый синдром (атаксия, интенционный тремор),
поражение глазодвигательных нервов (птоз, наружная офтальмоплегия),
полиневропатия,
инсультоподобные пароксизмы, мигренеподобные головные боли,
черепно-лицевая дисморфия,
дисметаболические проявления (рвота,эпизоды летаргии,комы),
дыхательные нарушения (апноэ, гипервентилляция, тахипноэ)
поражение сердца, печени, почек,
прогрессирующее течение заболевания.
,судороги (миоклонические")
Слайд 14Признаки поражения соединительной ткани (гипермобильный синдром, гиперэластичность кожи, нарушение осанки и
др.)
Нейродегенеративные проявления, лейкопатии на МРТ
Повторные эпизоды нарушения сознания или необъяснимые эпизоды рвоты у новорожденных
Необъяснимая атаксия
Отставание в умственном развитии без определенных причин ( John H.Menkes 2006,2007)
Семейный анамнез
Внезапное ухудшение состояния ребенка (судороги, рвота, расстройства дыхания, вялость, слабость, нарушения мышечного тонуса (чаще мышечная гипотония), кома, летаргия, поражение печени и почек, не поддающееся обычной терапии
Нейродегенеративные проявления, лейкопатии на МРТ
Повторные эпизоды нарушения сознания или необъяснимые эпизоды рвоты у новорожденных
Необъяснимая атаксия
Отставание в умственном развитии без определенных причин ( John H.Menkes 2006,2007)
Семейный анамнез
Внезапное ухудшение состояния ребенка (судороги, рвота, расстройства дыхания, вялость, слабость, нарушения мышечного тонуса (чаще мышечная гипотония), кома, летаргия, поражение печени и почек, не поддающееся обычной терапии
Нейродегенеративные проявления, лейкопатии на")
Слайд 15Биохимические тесты
лактат-пируват-ацидоз . НОРМАЛЬНЫЕ ПОКАЗАТЕЛИ МОЛОЧНОЙ КИСЛОТЫ НЕ ИСКЛЮЧАЮТ МИТОХОНДРИАЛЬНОГО
ЗАБОЛЕВАНИЯ [Chinnery P 2006]
кетоновые тела
ацилкарнитины плазмы, органические и аминокислоты крови и мочи.
Биопсия скелетных мышц с проведением специфических гистохимических реакций, выявление феномена “рваных красных волокон”. Синдромы с RRF – MELAS, MERRF, KSS,PEO(прогрессирующая наружная офтальмоплегия, синдром Пирсона. Синдромы без RRF – болезнь Лея,NARP, LHON (наследственная оптическая нейропатия Лебера)
Оценка содержания мышечного коэнзима Q10 у детей с дефектом ферментов дыхательной цепи митохондрий [Miles MV et all,2008]
Цитоморфоденситометрическое исследование активности митохондрий лимфоцитов – снижение количества, увеличение объема, снижение активности
Генетические исследования – определение частых мутаций, сиквенс митохондриальной ДНК
кетоновые тела
ацилкарнитины плазмы, органические и аминокислоты крови и мочи.
Биопсия скелетных мышц с проведением специфических гистохимических реакций, выявление феномена “рваных красных волокон”. Синдромы с RRF – MELAS, MERRF, KSS,PEO(прогрессирующая наружная офтальмоплегия, синдром Пирсона. Синдромы без RRF – болезнь Лея,NARP, LHON (наследственная оптическая нейропатия Лебера)
Оценка содержания мышечного коэнзима Q10 у детей с дефектом ферментов дыхательной цепи митохондрий [Miles MV et all,2008]
Цитоморфоденситометрическое исследование активности митохондрий лимфоцитов – снижение количества, увеличение объема, снижение активности
Генетические исследования – определение частых мутаций, сиквенс митохондриальной ДНК

Слайд 16НАИБОЛЕЕ ЧАСТО ВСТРЕЧАЮЩИЕСЯ В ДЕТСКОМ ВОЗРАСТЕ КЛИНИЧЕСКИЕ СИНДРОМЫ
Синдром MELAS – митохондриальная
энцефаломиопатия, лактат-ацидоз и инсультоподобные пароксизмы
Синдром MERRF – миоклонус-эпилепсия, рваные красные волокна
KSS – синдром Кернса-Сейра – характеризуется птозом, офтальмоплегией, пигментным ретинитом, атаксией, нарушением сердечного проведения
Синдром NARP – нейропатия,атаксия,пигментный ретинит
Синдром Лея – подострая некротизирующая энцефаломиелопатия
Болезнь Лебера – наследственная оптическая нейропатия
Синдром MERRF – миоклонус-эпилепсия, рваные красные волокна
KSS – синдром Кернса-Сейра – характеризуется птозом, офтальмоплегией, пигментным ретинитом, атаксией, нарушением сердечного проведения
Синдром NARP – нейропатия,атаксия,пигментный ретинит
Синдром Лея – подострая некротизирующая энцефаломиелопатия
Болезнь Лебера – наследственная оптическая нейропатия

Слайд 17Синдром Лея
Подострая некротизирующая энцефаломиелопатия – описана в 1951 году Denis Leigh
– у 6 мес младенца,развившего регресс психо-моторного развития и погибшего через 6 недель. На аутопсии выявлены множественные симметричные очаги губчатой дегенерации с микроваскулярной пролиферацией в таламусе,мозжечке,задних столбах спинного мозга,оптического нерва.
Дебют – часто острое начало, после перенесенного интеркуррентного заболевания, большинство пациентов – раннего возраста- до 2 лет,однако, некоторые пациенты – детского и подросткового возраста.
Клинические проявления – регресс развития, диффузная мышечная гипотония, проблемы вскармливания, прогрессирующая потеря зрения, потеря слуха, нистагм,атаксия,судороги, респираторные нарушения.
У детей старшего возраста – атаксия,мышечная дистония, нарушения интеллектуального развития, дизартрия,рвота,диаррея.
Диагностика- МРТ, лактат-пируват ацидоз,генетические исследования
Дебют – часто острое начало, после перенесенного интеркуррентного заболевания, большинство пациентов – раннего возраста- до 2 лет,однако, некоторые пациенты – детского и подросткового возраста.
Клинические проявления – регресс развития, диффузная мышечная гипотония, проблемы вскармливания, прогрессирующая потеря зрения, потеря слуха, нистагм,атаксия,судороги, респираторные нарушения.
У детей старшего возраста – атаксия,мышечная дистония, нарушения интеллектуального развития, дизартрия,рвота,диаррея.
Диагностика- МРТ, лактат-пируват ацидоз,генетические исследования

Слайд 18Болезнь Лея (подострая некротизирующая энцефаломиелопатия)
Мутации митохондриальных или ядерных генов, отвечающие за
возникновение болезни Лея.
Мутации митохондриальных или ядерных генов, отвечающие за возникновение болезни Лея.")
Слайд 19Синдром MERRF
Мультисистемное заболевание – основные характеристики
Миоклонус
Генерализованная эпилепсия
Мозжечковая атаксия
Рваные красные
волокна при мышечной биопсии
Дополнительные критерии
Психомоторный регресс
Мышечная слабость, миопатия
Аксональная периферическая нейропатия
нейросенсорная тугоухость
Низкий рост
лактат-ацидоз, гиперкетонемия;
Могут встречаться- атрофия зрительного нерва, кардиомиопатия, пигментная ретинопатия, множественный липоматоз
COX-негативные красные миофибриллы с рваными краями и SDH-позитивные миофибриллы;
A8344G и Т8356С мутация в гене тРНКLys мтДНК- 80% пациентов. MERRF ассоциирован примерно с 6 точковыми мутациями мтДНК
Дополнительные критерии
Психомоторный регресс
Мышечная слабость, миопатия
Аксональная периферическая нейропатия
нейросенсорная тугоухость
Низкий рост
лактат-ацидоз, гиперкетонемия;
Могут встречаться- атрофия зрительного нерва, кардиомиопатия, пигментная ретинопатия, множественный липоматоз
COX-негативные красные миофибриллы с рваными краями и SDH-позитивные миофибриллы;
A8344G и Т8356С мутация в гене тРНКLys мтДНК- 80% пациентов. MERRF ассоциирован примерно с 6 точковыми мутациями мтДНК

Слайд 20Синдром Кернса-Сейра
Описан в 1958 году у 2 пациентов с пигментным ретинитом,
наружной офтальмоплегией и нарушением внутрисердечного проведения.
Облигатные характеристики синдрома, определенные Lewis P.Rowland 1983
дебют до 20 летнего возраста,
пигментный ретинит
прогрессирующая наружная офтальмоплегия
Дополнительные – сердечные блокады
Мозжечковая атаксия
Белок в CMЖ – более 100 мг\dL.
Облигатные характеристики синдрома, определенные Lewis P.Rowland 1983
дебют до 20 летнего возраста,
пигментный ретинит
прогрессирующая наружная офтальмоплегия
Дополнительные – сердечные блокады
Мозжечковая атаксия
Белок в CMЖ – более 100 мг\dL.

Слайд 21У большинства пациентов встречаются
-низкий рост
-потеря слуха
-нарушение интеллектуального развития
-мышечная слабость
Начало заболевания
– птоз,офтальмопарез или оба эти состояния
Генетика – делеция мт ДНК у 90%, у некоторых пациентов может быть дупликация[Moraes CT et al 1989]
Генетика – делеция мт ДНК у 90%, у некоторых пациентов может быть дупликация[Moraes CT et al 1989]

Слайд 22МИТОХОНДРИАЛЬНАЯ ЭНЦЕФАЛОМИОПАТИЯ, ЛАКТАТ-АЦИДОЗ И ИНСУЛЬТОПОДОБНЫЕ ПАРОКСИЗМЫ (MELAS)
Мультисистемное заболевание, описанное в 1984
г Pavlakis SG et all, характеризующееся
- Инсультоподобными пароксизмами,возникающими в молодом возрасте (до 40 лет) У более 60% пациентов заболевание дебютирует до 15 лет.
Энцефалопатией, характеризующейся судорогами, деменцией
Митохондриальной миопатией с лактат-ацидозом,рваными красными волокнами(волокнами(COX- позитивные красные миофибриллы с рваными краями, SDH-позитивные миофибриллы и SDH-реактивные кровеносные сосуды в мышечном биоптате)(сочетанная недостаточность комплексов дыхательной цепи митохондрий), гиперкетонемия
Дополнительные проявления: низкий рост,кардиомиопатия, кальцинаты базальных ганглиев, миоклонус,атаксия,мигренеподобные головные боли,атрофия зрительных нервов, пигментная ретинопатия,потеря слуха, офтальмоплегия, диабет,нарушение сердечной проводимости,гастроинтестинальные нарушения,нефропатия,тошнота
Основная характеристика этого заболевания – инсультоподобные пароксизмы,наиболее часто локализующиеся в затылочной области, приводящие к гемианопсии или корковой слепоте. Однако, зоны могут быть различными.
- Инсультоподобными пароксизмами,возникающими в молодом возрасте (до 40 лет) У более 60% пациентов заболевание дебютирует до 15 лет.
Энцефалопатией, характеризующейся судорогами, деменцией
Митохондриальной миопатией с лактат-ацидозом,рваными красными волокнами(волокнами(COX- позитивные красные миофибриллы с рваными краями, SDH-позитивные миофибриллы и SDH-реактивные кровеносные сосуды в мышечном биоптате)(сочетанная недостаточность комплексов дыхательной цепи митохондрий), гиперкетонемия
Дополнительные проявления: низкий рост,кардиомиопатия, кальцинаты базальных ганглиев, миоклонус,атаксия,мигренеподобные головные боли,атрофия зрительных нервов, пигментная ретинопатия,потеря слуха, офтальмоплегия, диабет,нарушение сердечной проводимости,гастроинтестинальные нарушения,нефропатия,тошнота
Основная характеристика этого заболевания – инсультоподобные пароксизмы,наиболее часто локализующиеся в затылочной области, приводящие к гемианопсии или корковой слепоте. Однако, зоны могут быть различными.
Мультисистемное заболевание, описанное в 1984 г Pavlakis SG et")
Слайд 24ОСОБЕННОСТИ ИНСУЛЬТА ПРИ СИНДРОМЕ MELAS
Частая локализация в затылочной области, приводящая к
гемианопсии или корковой слепоте.
Инсультоподобные пароксизмы атипичные, т.е. поражают в основном молодых , часто провоцируются заболеваниями, сопровождающимися фебрильной температурой, мигренеподобной головной болью, возникают после судорог
Очаги часто лежат вне региона крупных церебральных артерий, чаще поражая кору или глубинные структуры белого вещества головного мозга (Mathews PM et al 1991)
Острые MELAS очаги могут флюктуировать,мигрировать или даже исчезать. Кальцификаты базальных ганглиев – частая находка (Sue SM et al, 1998)
Ангиография подтверждает отсутствие патологии со стороны крупных сосудов (Hirano M,Pavlakis S 1994).
Инсультоподобные пароксизмы атипичные, т.е. поражают в основном молодых , часто провоцируются заболеваниями, сопровождающимися фебрильной температурой, мигренеподобной головной болью, возникают после судорог
Очаги часто лежат вне региона крупных церебральных артерий, чаще поражая кору или глубинные структуры белого вещества головного мозга (Mathews PM et al 1991)
Острые MELAS очаги могут флюктуировать,мигрировать или даже исчезать. Кальцификаты базальных ганглиев – частая находка (Sue SM et al, 1998)
Ангиография подтверждает отсутствие патологии со стороны крупных сосудов (Hirano M,Pavlakis S 1994).

Слайд 25Количественный анализ церебрального кровотока, исследованный с помощью ксенон КТ выявил нормальный
или усиленный кровоток в зоне острого повреждения и непораженных областях, что предполагает, что инсультоподобные проявления возникают не из-за фокальной ишемии(Morita K, et.al 1989, Ooiwa Y et al.1993)
SPECT обычно выявляет снижение аккумуляции вводимого вещества в зонах повреждения, возможно, в связи с потерей метаболически активных клеток(Saton M et al 1991, Watanabe Y et al 1998)
Имеются интересные наблюдения об увеличении поглощения вещества при SPECT и увеличение регионального кровотока при PET исследованиях за 3-6 дней до появления инсульта (Takahashi S et al 1998). Возможно, что повышение кровотока до острого инсультоподобного эпизода может отражать локализованный ответ на повышение уровня лактата,фокального повышения метаболизма или повреждения вазорегуляции.
MRS (магниторезонансная спектроскопия) выявляет повышение лактата в головном мозга, отражающее компенсаторное повышение анаэробного гликолиза. Выявлено также, что содержание вентрикулярного лактата находится в прямой корреляционной зависимости от неврологическогодефицита при MELAS (Dubeau F et al 2000, Kaufmann P et al 2004)
SPECT обычно выявляет снижение аккумуляции вводимого вещества в зонах повреждения, возможно, в связи с потерей метаболически активных клеток(Saton M et al 1991, Watanabe Y et al 1998)
Имеются интересные наблюдения об увеличении поглощения вещества при SPECT и увеличение регионального кровотока при PET исследованиях за 3-6 дней до появления инсульта (Takahashi S et al 1998). Возможно, что повышение кровотока до острого инсультоподобного эпизода может отражать локализованный ответ на повышение уровня лактата,фокального повышения метаболизма или повреждения вазорегуляции.
MRS (магниторезонансная спектроскопия) выявляет повышение лактата в головном мозга, отражающее компенсаторное повышение анаэробного гликолиза. Выявлено также, что содержание вентрикулярного лактата находится в прямой корреляционной зависимости от неврологическогодефицита при MELAS (Dubeau F et al 2000, Kaufmann P et al 2004)

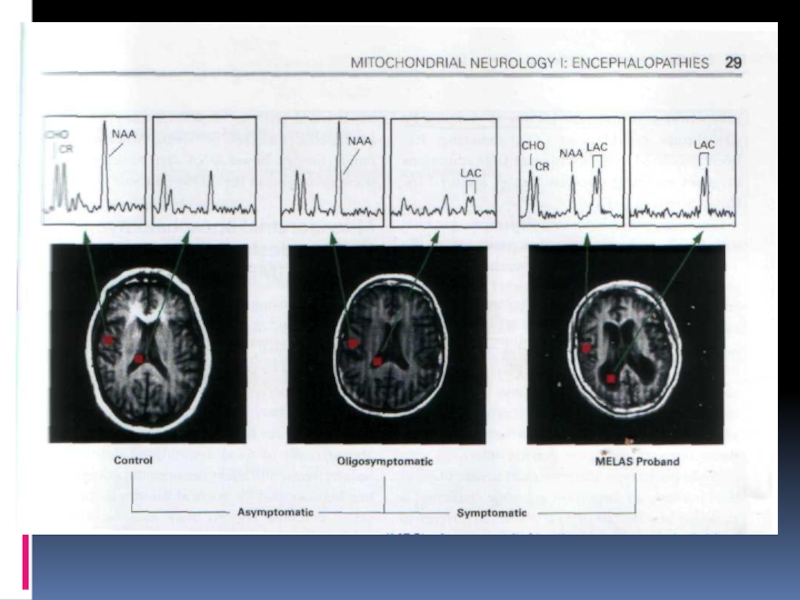
Слайд 27
Т.О. суммируя все вышеизложенное, нейрорадиологические исследования при синдроме MELAS выявили, что
атипичные инсультоподобные пароксизмы возникают не столько от острой ишемии, сколько от метаболической дисфункции со снижением окислительного фосфорилирования,повышением уровня лактата и снижением синтеза АТФ [ DiMauro S. et al 2006].
Это доказывает необходимость дифференцированного подхода в терапии данных состояний, которые могут мимикрировать под ишемический инсульт [Finsterer J 2009].
Несмотря на интенсивное изучение патогенеза инсультоподобных пароксизмов при синдроме MELAS, он остается неясным. Однако, предложены 2 основные теории : ишемическая васкулярная – “митохондриальная ангиопатия” и общая цитопатическая гипотеза, причиной которой является “митохондриальная цитопатия” [Iizuka t., Sakai F 2005]. Однако, большинство исследователей придерживается теории, что инсультоподобные пароксизмы являются неишемическим нейроваскулярным событием [Iizuka T et al 2007].
Дифференциальная диагностика – коагулопатии, антифосфолипидный синдром,тромбофилии,синдром Эллерса-Данлоса 1Y типа, гомоцистинурия
Это доказывает необходимость дифференцированного подхода в терапии данных состояний, которые могут мимикрировать под ишемический инсульт [Finsterer J 2009].
Несмотря на интенсивное изучение патогенеза инсультоподобных пароксизмов при синдроме MELAS, он остается неясным. Однако, предложены 2 основные теории : ишемическая васкулярная – “митохондриальная ангиопатия” и общая цитопатическая гипотеза, причиной которой является “митохондриальная цитопатия” [Iizuka t., Sakai F 2005]. Однако, большинство исследователей придерживается теории, что инсультоподобные пароксизмы являются неишемическим нейроваскулярным событием [Iizuka T et al 2007].
Дифференциальная диагностика – коагулопатии, антифосфолипидный синдром,тромбофилии,синдром Эллерса-Данлоса 1Y типа, гомоцистинурия

Слайд 28ЛЕЧЕНИЕ МИТОХОНДРИАЛЬНЫХ БОЛЕЗНЕЙ
Нет достоверных подтверждений, что какая-либо терапия может быть эффективна
в отношении митохондриальных болезней. Различные антиоксиданты, витамины, кофакторы дыхательной цепи применяются у детей, страдающих обменными расстройствами.
При синдроме MELAS лечение должно быть направлено на лечение судорог, эндокринных расстройств,устранение последствий инсульта.
Т.к. уровень лактата часто коррелирует с тяжестью неврологических проявлений, целесообразно применять дихлорацетат (у нас в стране используется демифосфон) для снижения уровня лактата (Kaufmann P et al, 2006)
В исследованиях японских авторов Koga Y et al,2002, 2005,2008 использовался в\в L-аргинин,предшественник NO, для стимуляции вазодилятации в остром периоде инсульта с хорошими результатами, а также пероральное его применение для снижения тяжести последующих эпизодов
Казанцева Л.З., Юрьева Э.А., Николаева Е.А. и др 2001-Основные методы лечения детей, страдающих митохондриальными заболеваниями
При синдроме MELAS лечение должно быть направлено на лечение судорог, эндокринных расстройств,устранение последствий инсульта.
Т.к. уровень лактата часто коррелирует с тяжестью неврологических проявлений, целесообразно применять дихлорацетат (у нас в стране используется демифосфон) для снижения уровня лактата (Kaufmann P et al, 2006)
В исследованиях японских авторов Koga Y et al,2002, 2005,2008 использовался в\в L-аргинин,предшественник NO, для стимуляции вазодилятации в остром периоде инсульта с хорошими результатами, а также пероральное его применение для снижения тяжести последующих эпизодов
Казанцева Л.З., Юрьева Э.А., Николаева Е.А. и др 2001-Основные методы лечения детей, страдающих митохондриальными заболеваниями

Слайд 29ИСПОЛЬЗУЕМЫЕ ПРЕПАРТЫ
КоэнзимQ10 – от 90 до 200 мг/сут
Lкарнитин – от
10 мг до 1-2 г/сут
Вит В1 400 мг/сут
Вит В2 100 мг/сут
Вит С до 1 г/сут
Вит Е 400 МЕ в сут
Никотинамид до 500 мг/сут
Янтарная кислота от 25 мг до 1.5 г в сутки
Демифосфон 15% 1 мл на 5 кг веса
Цитохром С 4.0 в,в,
Реамберин в/в,
цитофлавин в/в и перорально
Вит В1 400 мг/сут
Вит В2 100 мг/сут
Вит С до 1 г/сут
Вит Е 400 МЕ в сут
Никотинамид до 500 мг/сут
Янтарная кислота от 25 мг до 1.5 г в сутки
Демифосфон 15% 1 мл на 5 кг веса
Цитохром С 4.0 в,в,
Реамберин в/в,
цитофлавин в/в и перорально

Слайд 30Кортикостероиды, минералокортикоиды - при развитии надпочечниковой недостаточности
Антиконвульсанты (исключая вальпроевую кислоту и
ее производные, ограничивая барбитураты)
В наших исследованиях наиболее эффективной противосудорожной терапией являлось использование препарата леветирацетам, топирамат или их сочетаний
В наших исследованиях наиболее эффективной противосудорожной терапией являлось использование препарата леветирацетам, топирамат или их сочетаний
В")
Слайд 31Имеется большой пул заболеваний, причиной которых является не мутации митохондриальной ДНК,
а мутации ядерной ДНК, кодирующей работу митохондрий. К ним относятся
Болезнь Барта – миопатия,кардиомиопатия, транзиторные нейтро и тромбоцитопении
Митохондриальная гастроинтестинальная энцефалопатия (аутосомно-рецессивное мультисистемное заболевание)-птоз,офтальмоплегия,периферическая нейропатия, гастроинтестинальная дисфункция, приводящая к кахексии, лейкоэнцефалопатия. Дебют от периода новорожденности до 43 лет
Синдром Лея
Болезнь Барта – миопатия,кардиомиопатия, транзиторные нейтро и тромбоцитопении
Митохондриальная гастроинтестинальная энцефалопатия (аутосомно-рецессивное мультисистемное заболевание)-птоз,офтальмоплегия,периферическая нейропатия, гастроинтестинальная дисфункция, приводящая к кахексии, лейкоэнцефалопатия. Дебют от периода новорожденности до 43 лет
Синдром Лея

Слайд 32Важность своевременной диагностики митохондриальных болезней, поиска клинических и параклинических критериев этих
заболеваний на этапе предварительном, догенетическом, необходимо для подбора адекватной метаболической терапии и предотвращения ухудшения состояния или инвалидизации больных с этими редкими заболеваниями.







