- Главная
- Разное
- Дизайн
- Бизнес и предпринимательство
- Аналитика
- Образование
- Развлечения
- Красота и здоровье
- Финансы
- Государство
- Путешествия
- Спорт
- Недвижимость
- Армия
- Графика
- Культурология
- Еда и кулинария
- Лингвистика
- Английский язык
- Астрономия
- Алгебра
- Биология
- География
- Детские презентации
- Информатика
- История
- Литература
- Маркетинг
- Математика
- Медицина
- Менеджмент
- Музыка
- МХК
- Немецкий язык
- ОБЖ
- Обществознание
- Окружающий мир
- Педагогика
- Русский язык
- Технология
- Физика
- Философия
- Химия
- Шаблоны, картинки для презентаций
- Экология
- Экономика
- Юриспруденция
Болезни клеточных органелл презентация
Содержание
- 1. Болезни клеточных органелл
- 2. 1.Митохондриальные болезни
- 3. Митохондрии Митохондриальная ДНК
- 4. Схема строения митохондриальной ДНК
- 5. Углеводы Жиры глюкоза лактат
- 7. Клинические проявления митохондриальных болезней (основные) миопатический синдром
- 8. Клинические проявления митохондриальных болезней (дополнительные) снижение слуха
- 9. Схема строения митохондрии
- 10. Феномен RRF (рваных (или шероховатых) красных волокон) в биоптатах мышц при митохондриальных болезнях.
- 12. Синдром Кернса-Сейра
- 13. Синдром Кернса-Сейра Манифестация с 4 - 18
- 14. Динамика появления клинических симптомов Частичный птоз век
- 15. Причина синдрома Кернса-Сейра - делеция митохондриальной ДНК
- 16. Делеция митохондриальной ДНК
- 17. Клиническая экспрессия митохондриальной делеции зависит от Локализации
- 18. Синдром MELAS - (митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные
- 19. Синдром MERRF (миоклонус-эпилепсия с «рваными» красными волокнами).
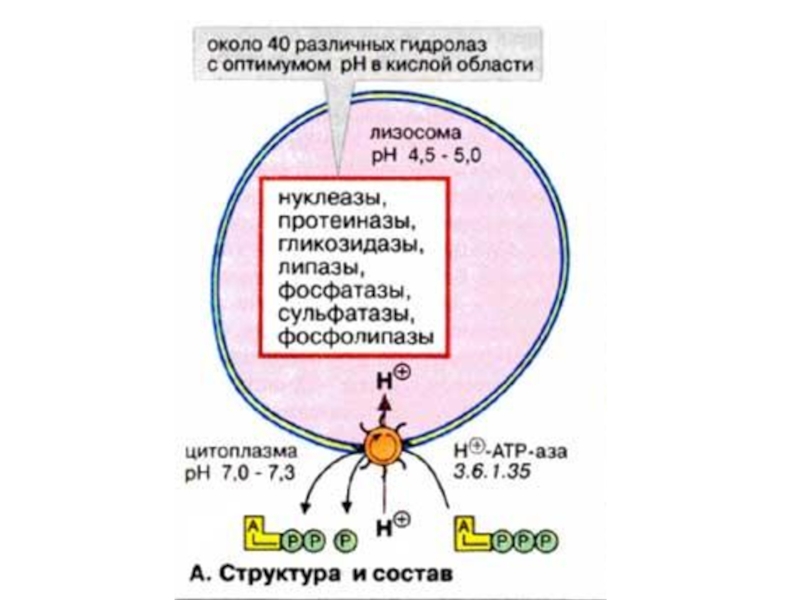
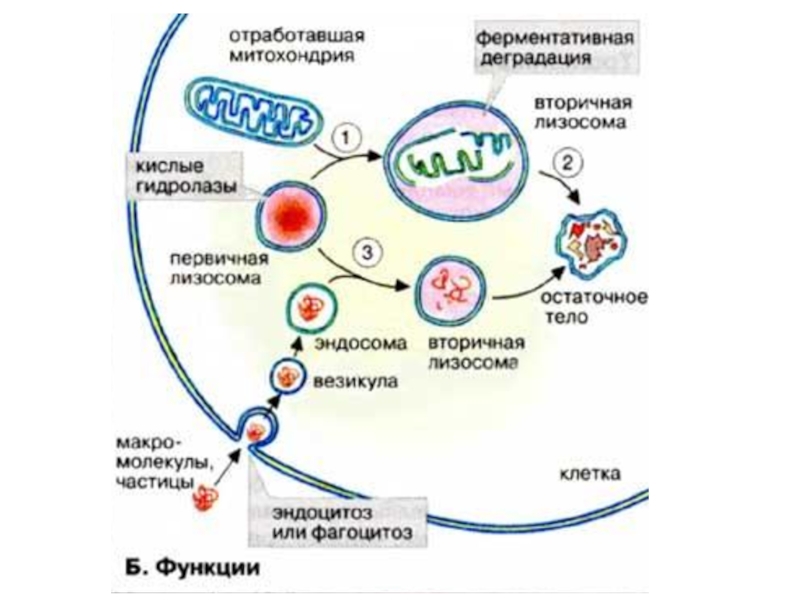
- 20. 2.Лизосомные болезни накопления
- 23. Классификация лизосомных болезней мукополисахаридозы сфинголипидозы муколипидозы
- 24. Мукополисахаридозы Мукополисахариды являются сложными гетерогенными соединениями. Из
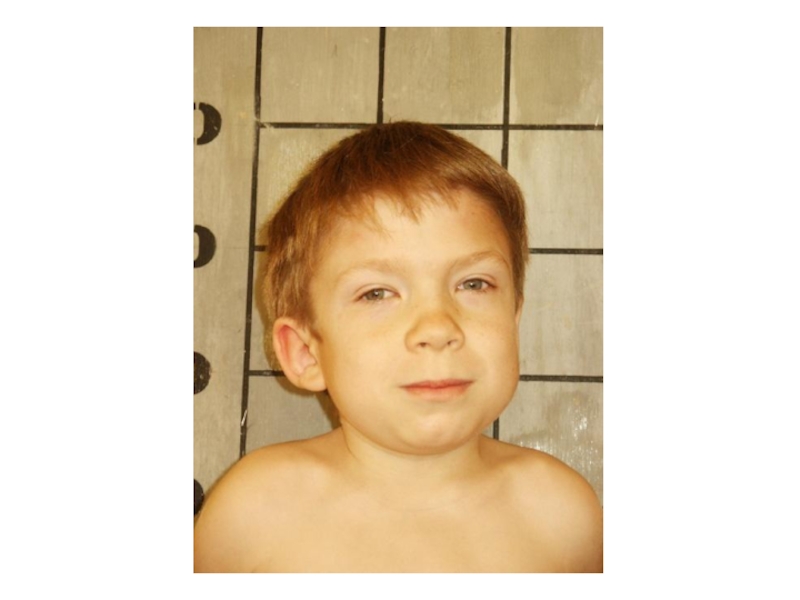
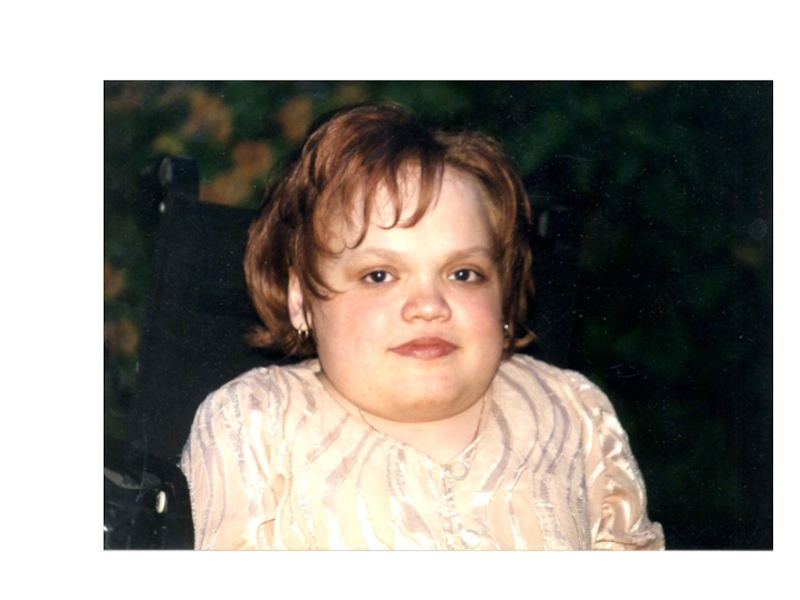
- 25. Лицо больного мукополисахаридозом
- 26. Типы мукополисахаридозов I тип
- 27. Синдром Гурлер (МПСI)
- 28. Синдром Гурлер (МПСI) Ген локализован в 22q11.
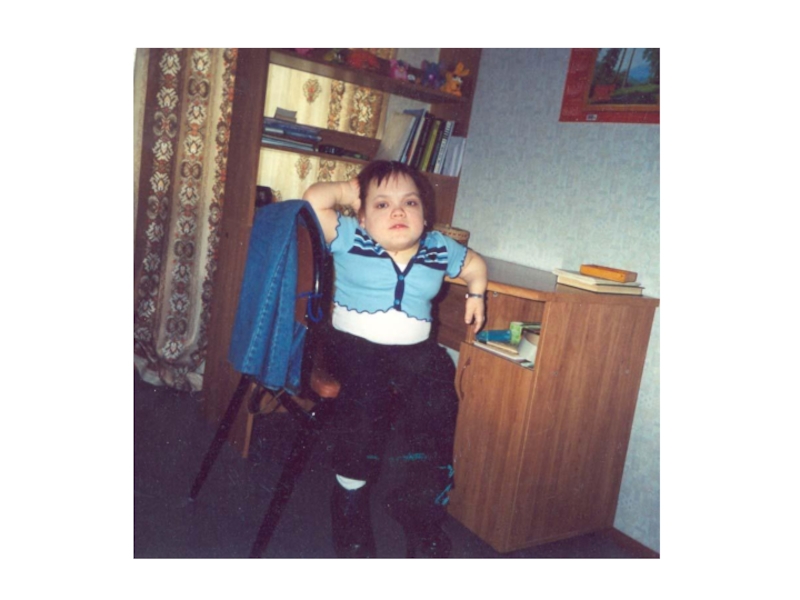
- 29. Гипертрихоз
- 30. Рентгенологические изменения при синдроме Гурлер:
- 31. Синдром Гурлер-Шейе
- 35. Синдром Хантера (МПСII)
- 36. Синдром Хантера (МПСII) Ген локализован в Хq27.1-q28.
- 39. Ферментозаместительная терапия больных с МПСI и МПСII.
- 40. Эффективность лечения ферментозамещающими препаратами «Альдуразим» и «Элапраза»
- 41. Синдром Моркио (МПСIV)
- 42. Рентгенологические признаки синдрома Моркио: деформация костей таза,
- 46. Морато-Лами синдром (МПСVI)
- 47. 3.Пероксисомные болезни
- 48. Адренолейкодистрофия Дегенеративное заболевание белого вещества головного мозга
- 49. МРТ больного с адренолейкодистрофией




Слайд 5
Углеводы
Жиры
глюкоза
лактат
жирные кислоты
Ацетил КоА
Цикл Кребса
Бета-окисление
I
II
III
IV
V
АТФ
Дыхательная цепь
L-карнитин
рибофлавин
никотинамид
рибофлавин
рибофлавин
тиамин биотин
Коэнзим Q10
М и т о х о н д р и я

Слайд 7Клинические проявления митохондриальных болезней (основные)
миопатический синдром - слабость и атрофия мышц,
снижение мышечного тонуса, мышечные боли (“крампи”), непереносимость физической нагрузки (усиление мышечной слабости и боли, появление рвоты и головной боли);
поражение нервной системы - нарушение психического и статико-моторного развития, регресс приобретенных навыков, судороги, в т.ч. тонико-клонические, миоклонические, атаксия, спастичность, птоз, наружная офтальмоплегия; в старшем возрасте инсультоподобные эпизоды, головные боли, головокружение, периферическая нейропатия;
поражение сердца в виде гипертрофической или дилатационной кардиомиопатии, блокады проводящей системы сердца;
поражение нервной системы - нарушение психического и статико-моторного развития, регресс приобретенных навыков, судороги, в т.ч. тонико-клонические, миоклонические, атаксия, спастичность, птоз, наружная офтальмоплегия; в старшем возрасте инсультоподобные эпизоды, головные боли, головокружение, периферическая нейропатия;
поражение сердца в виде гипертрофической или дилатационной кардиомиопатии, блокады проводящей системы сердца;
миопатический синдром - слабость и атрофия мышц, снижение мышечного тонуса, мышечные")
Слайд 8Клинические проявления митохондриальных болезней (дополнительные)
снижение слуха сенсоневрального происхождения;
нарушение зрения - атрофия
зрительных нервов, пигментная дегенерация сетчатки, катаракта, помутнение роговицы;
поражение печени - прогрессирующее увеличение печени с нарушением функции и развитием печеночной недостаточности;
поражение почек (фосфатурия, глюкозурия, аминоацидурия);
эндокринные нарушения - задержка роста и костного возраста, нарушение полового развития, гипогликемия, сахарный и несахарный диабет, гипоталамо-гипофизарная недостаточность, дефицит гормона роста, гипотиреоз, гипопаратиреоз, гиперальдостеронизм;
желудочно-кишечные расстройства - повторные рвоты (особенно после физической нагрузки), поносы с признаками недостаточности поджелудочной железы.
поражение печени - прогрессирующее увеличение печени с нарушением функции и развитием печеночной недостаточности;
поражение почек (фосфатурия, глюкозурия, аминоацидурия);
эндокринные нарушения - задержка роста и костного возраста, нарушение полового развития, гипогликемия, сахарный и несахарный диабет, гипоталамо-гипофизарная недостаточность, дефицит гормона роста, гипотиреоз, гипопаратиреоз, гиперальдостеронизм;
желудочно-кишечные расстройства - повторные рвоты (особенно после физической нагрузки), поносы с признаками недостаточности поджелудочной железы.
снижение слуха сенсоневрального происхождения;нарушение зрения - атрофия зрительных нервов, пигментная дегенерация")

Слайд 10Феномен RRF (рваных (или шероховатых) красных волокон) в биоптатах мышц при
митохондриальных болезнях.
 красных волокон) в биоптатах мышц при митохондриальных болезнях.")

Слайд 13Синдром Кернса-Сейра
Манифестация с 4 - 18 лет
Снижение толерантности к физ. нагрузке,
миопатич. синдром, птоз, офтальмоплегия, пигментный ретинит, нарушение сердечной проводимости, атаксия, глухота, сахарный диабет, низкий рост, умственная отсталость
Делеция мит.ДНК у 85-95 % больных
Делеция мит.ДНК у 85-95 % больных

Слайд 14Динамика появления клинических симптомов
Частичный птоз век - 9 лет
Частичная офтальмоплегия -
10,5 лет
Снижение толерантности к физической нагрузке - 11 лет
Снижение толерантности к физической нагрузке - 11 лет

Слайд 15Причина синдрома Кернса-Сейра - делеция митохондриальной ДНК
Размер от 1,3 до 8
тыс. пар нуклеотидов
Наиболее частая делеция: размер 4,9 тыс. пар нуклеотидов
Наиболее частая делеция: размер 4,9 тыс. пар нуклеотидов


Слайд 17Клиническая экспрессия митохондриальной делеции зависит от
Локализации делеции в тканях
Чувствительности данной ткани
к энергетическому дефициту
Уровня гетероплазмии (% соотношение мутантной и нормальной ДНК) в клетке и ткани
Уровня гетероплазмии (% соотношение мутантной и нормальной ДНК) в клетке и ткани

Слайд 18Синдром MELAS -
(митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные эпизоды).
В основе заболевания – точковая
мутация митохондриального гена транспортной РНК.
Тип наследования материнский.
Клинические проявления: непереносимость физ. нагрузки, мышечная слабость, судороги, тошнота, сонливость, головные боли, инсультоподобные состояния, и, как следствие последних, - очаговая неврологическая симптоматика, деменция, глухота, снижение зрения, низкий рост.
Тип наследования материнский.
Клинические проявления: непереносимость физ. нагрузки, мышечная слабость, судороги, тошнота, сонливость, головные боли, инсультоподобные состояния, и, как следствие последних, - очаговая неврологическая симптоматика, деменция, глухота, снижение зрения, низкий рост.
.В основе заболевания – точковая мутация митохондриального гена транспортной")
Слайд 19Синдром MERRF
(миоклонус-эпилепсия с «рваными» красными волокнами).
В основе заболевания – точковая мутация
митохондриального гена транспортной РНК.
Тип наследования материнский.
Клинические проявления: Генерализованные тонико-клонические судороги, миоклонии, мышечная слабость, судороги, атаксия, деменция, глухота, снижение зрения, низкий рост.
Тип наследования материнский.
Клинические проявления: Генерализованные тонико-клонические судороги, миоклонии, мышечная слабость, судороги, атаксия, деменция, глухота, снижение зрения, низкий рост.
.В основе заболевания – точковая мутация митохондриального гена транспортной РНК.")


Слайд 24Мукополисахаридозы
Мукополисахариды являются сложными гетерогенными соединениями. Из них хондроитин-серная и гиалуроновые кислоты
служат основными строительными элементами соединительной ткани.
Мукополисахаридозы – группа заболеваний, обусловленных генетическим дефектом ферментного расщепления углеводной части молекулы мукополисахаридов, при этом наблюдается накопление мукополисахаридов в тканях.
Мукополисахаридозы – группа заболеваний, обусловленных генетическим дефектом ферментного расщепления углеводной части молекулы мукополисахаридов, при этом наблюдается накопление мукополисахаридов в тканях.


Слайд 26Типы мукополисахаридозов
I тип — 1:20000 — 1:25000. Наследование аутосомно-рецессивное.
Дефицит α –L – идуронидазы.
тип I-H, синдром Гурлер.
тип I-S, болезнь Шейе
II тип — синдром Хантера. 1: 70 000. Наследование рецессивное, сцепленное с полом.
III тип — синдром Санфилиппо. 1 на 100 000—200 000. Наследование аутосомно-рецессивное.
Тип IIIА – дефицит сульфаминидазы.
Тип IIIВ – дефицит N-ацетил – α –D – глюкозаминидазы.
Тип IIIС – дефицит ацетил – КоА - α – глюкозаминидазы – N – ацетилтрансферазы.
Тип III D – дефицит N – ацетил – глюкозамин - 6 - сульфат – сульфатазы.
IV тип — синдром Моркио. 1: 40 000. Наследование аутосомно-рецессивное. Тип IV А - дефицит галактозамин-6-сульфатазы
Тип IV В - дефицит β-галактозидазы.
VI тип — синдром Марото—Лами. Наследование аутосомно-рецессивное. Дефицит арилсульфатазы В.
VII тип - синдром Слая. Наследование аутосомно-рецессивное. Дефицит β-глюкуронидазы.
VII тип - синдром Ди Ферранте. Наследование аутосомно-рецессивное.
тип I-H, синдром Гурлер.
тип I-S, болезнь Шейе
II тип — синдром Хантера. 1: 70 000. Наследование рецессивное, сцепленное с полом.
III тип — синдром Санфилиппо. 1 на 100 000—200 000. Наследование аутосомно-рецессивное.
Тип IIIА – дефицит сульфаминидазы.
Тип IIIВ – дефицит N-ацетил – α –D – глюкозаминидазы.
Тип IIIС – дефицит ацетил – КоА - α – глюкозаминидазы – N – ацетилтрансферазы.
Тип III D – дефицит N – ацетил – глюкозамин - 6 - сульфат – сульфатазы.
IV тип — синдром Моркио. 1: 40 000. Наследование аутосомно-рецессивное. Тип IV А - дефицит галактозамин-6-сульфатазы
Тип IV В - дефицит β-галактозидазы.
VI тип — синдром Марото—Лами. Наследование аутосомно-рецессивное. Дефицит арилсульфатазы В.
VII тип - синдром Слая. Наследование аутосомно-рецессивное. Дефицит β-глюкуронидазы.
VII тип - синдром Ди Ферранте. Наследование аутосомно-рецессивное.

")
Слайд 28Синдром Гурлер (МПСI)
Ген локализован в 22q11.
Клинические признаки:
Тяжелая симптоматика и ранняя
манифестация.
Грубые черты лица (гаргоилизм), гирсутизм, низкий рост, тугоподвижность суставов, кифозы, грыжи, гепатоспленомегалия, гипертрофическая кардиомиопатия, грубое снижение интеллекта, помутнение роговицы и глаукома, тугоухость.
Грубые черты лица (гаргоилизм), гирсутизм, низкий рост, тугоподвижность суставов, кифозы, грыжи, гепатоспленомегалия, гипертрофическая кардиомиопатия, грубое снижение интеллекта, помутнение роговицы и глаукома, тугоухость.
Ген локализован в 22q11.Клинические признаки: Тяжелая симптоматика и ранняя манифестация.Грубые черты лица (гаргоилизм),")

Слайд 30
Рентгенологические изменения при синдроме Гурлер:
Кубовидной формы позвонки с закругленными контурами,
углообразный кифоз пояснично-грудного отдела, гипоплазия концевых фаланг, остальные фаланги и пястные кости - короткие и широкие, таз «сдавлен» с боков.




")
Слайд 36Синдром Хантера (МПСII)
Ген локализован в Хq27.1-q28.
Клинические признаки:
Признаки подобные синдрому
Гурлер, но более поздняя манифестация, чем при синдроме Гурлер, менее выраженным нарушением интеллекта, отсутствием помутнения роговицы.
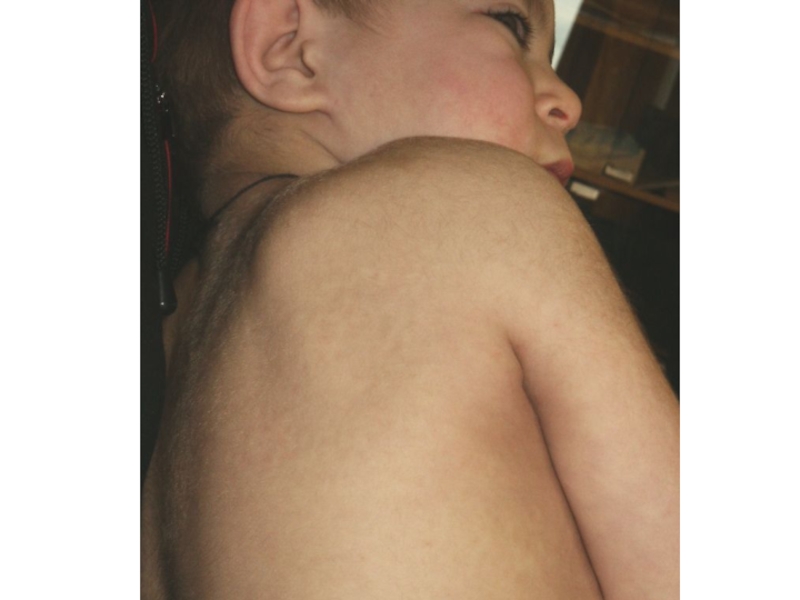
Узелково-папулезное поражение кожи в области лопаток, плеч и бедер (отложение липидов и мукополисахаридов).
Узелково-папулезное поражение кожи в области лопаток, плеч и бедер (отложение липидов и мукополисахаридов).
Ген локализован в Хq27.1-q28. Клинические признаки: Признаки подобные синдрому Гурлер, но более поздняя")
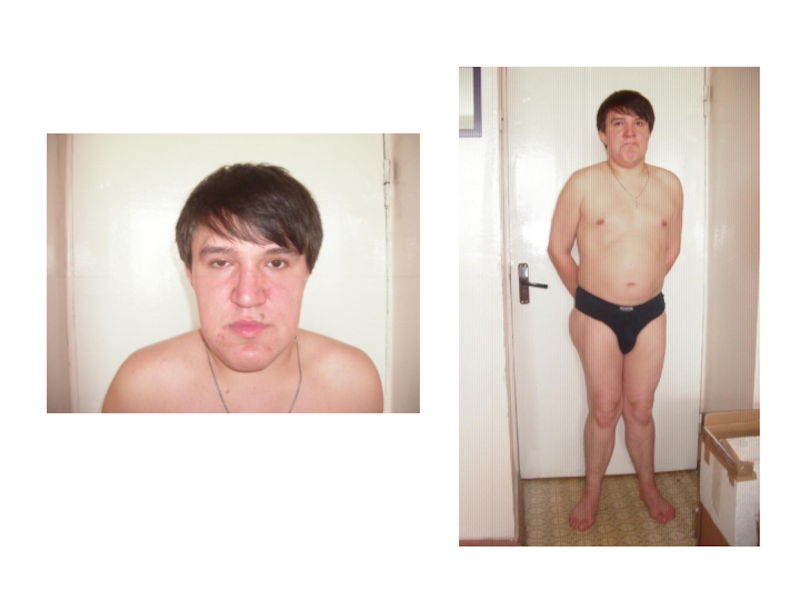
Слайд 39Ферментозаместительная терапия больных с МПСI и МПСII.
Альдуразим разработан американской фирмой
«Джинзайм» (GENZYME) для лечения МПСI
Элапраза разработана британско--американской компанией Шайер (SHIRE) для лечения МПСII
Ферменты вводятся внутривенно капельно в течение нескольких часов пожизненно один раз в неделю.
Элапраза разработана британско--американской компанией Шайер (SHIRE) для лечения МПСII
Ферменты вводятся внутривенно капельно в течение нескольких часов пожизненно один раз в неделю.
 для лечения")
Слайд 40Эффективность лечения ферментозамещающими препаратами «Альдуразим» и «Элапраза»
Улучшение общего состояния больных:
- нарастание двигательной активности
- увеличение длины тела
- уменьшение тугоподвижности суставов
- сокращение размеров сердца, печени и селезенки
- повышение концентрации внимания
- увеличение словарного запаса
- прекращение апноэ
- улучшение показателей функции внешнего
дыхания
- снижение показателей почечной экскреции
гликозаминогликанов (ГАГ)
- увеличение длины тела
- уменьшение тугоподвижности суставов
- сокращение размеров сердца, печени и селезенки
- повышение концентрации внимания
- увеличение словарного запаса
- прекращение апноэ
- улучшение показателей функции внешнего
дыхания
- снижение показателей почечной экскреции
гликозаминогликанов (ГАГ)

")
Слайд 42Рентгенологические признаки синдрома Моркио: деформация костей таза, вертлужных впадин, гипоплазия головок бедренных
костей, деформация костей кистей.


")

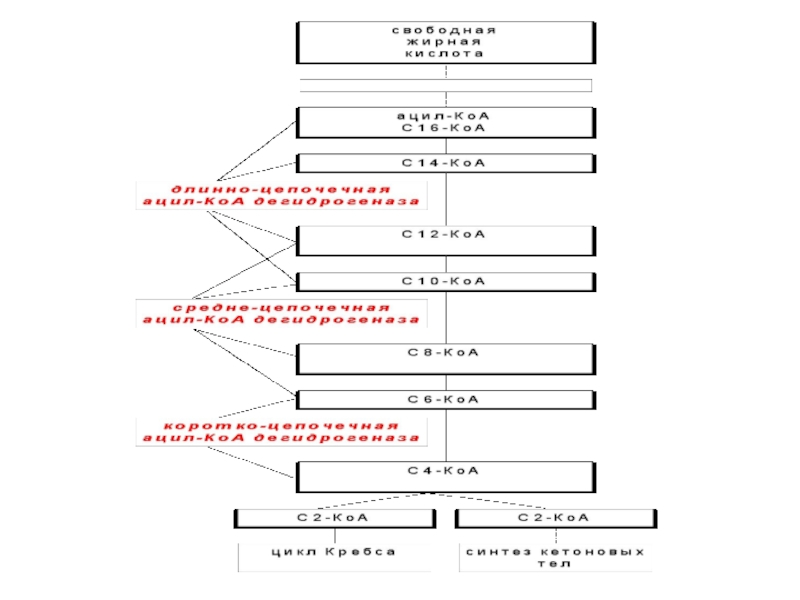
Слайд 48Адренолейкодистрофия
Дегенеративное заболевание белого вещества головного мозга и надпочечников, обусловленное дефектом обмена
жирных кислот.
Тип наследования рецессивный, сцепленный с Х-хромосомой. Ген ALD. Заболевание поражает преимущественно мальчиков (частота 1:20 000 рождений).
Выражены признаки недостаточности надпочечников (гиперпигментация кожи, гипогонадизм), сочетающиеся с интеллектуальная , поведенческая недостаточность (лабильность настроения, эйфория, депрессия), расстройства памяти, нарушение походки, постепенное развитие спастичности.
Атрофия зрительных нервов.
Фокальные приступы судорог.
Повышено содержание жирных кислот с длинной цепью.
Тип наследования рецессивный, сцепленный с Х-хромосомой. Ген ALD. Заболевание поражает преимущественно мальчиков (частота 1:20 000 рождений).
Выражены признаки недостаточности надпочечников (гиперпигментация кожи, гипогонадизм), сочетающиеся с интеллектуальная , поведенческая недостаточность (лабильность настроения, эйфория, депрессия), расстройства памяти, нарушение походки, постепенное развитие спастичности.
Атрофия зрительных нервов.
Фокальные приступы судорог.
Повышено содержание жирных кислот с длинной цепью.







