- Главная
- Разное
- Дизайн
- Бизнес и предпринимательство
- Аналитика
- Образование
- Развлечения
- Красота и здоровье
- Финансы
- Государство
- Путешествия
- Спорт
- Недвижимость
- Армия
- Графика
- Культурология
- Еда и кулинария
- Лингвистика
- Английский язык
- Астрономия
- Алгебра
- Биология
- География
- Детские презентации
- Информатика
- История
- Литература
- Маркетинг
- Математика
- Медицина
- Менеджмент
- Музыка
- МХК
- Немецкий язык
- ОБЖ
- Обществознание
- Окружающий мир
- Педагогика
- Русский язык
- Технология
- Физика
- Философия
- Химия
- Шаблоны, картинки для презентаций
- Экология
- Экономика
- Юриспруденция
Геномный анализ презентация
Содержание
- 1. Геномный анализ
- 2. ОБЩАЯ СТРАТЕГИЯ ИССЛЕДОВАНИЯ МАКРОМОЛЕКУЛ Получение ОБЪЕКТА исследования
- 3. Геномный анализ Геномный анализ — это один из цитогенетических
- 4. Методы выделения и очистки геномной ДНК
- 5. Выделение и очистка ДНК Основная цель
- 15. Существует несколько методов ДНК–тестирования (анализа) Электрофорез ДНК
- 16. Электрофорез – метод разделения макромолекул, различающихся по
- 17. Фракционирование НК. Гель-электрофорез ДНК
- 18. Преимущества агарозного геля прочность агарозного геля; крупнопористость
- 19. Агароза очень хрупка, легко разрушается при манипулировании.
- 20. При проведении электрофореза фрагменты ДНК мигрирют в
- 21. Разделение фрагментов ДНК происходит из-за наличия у
- 22. Компоненты электрофореза в агарозном геле Агароза Буфер для электрфореза Маркерная ДНК
- 23. В зависимости от выбранной процентности агарозы статистический
- 24. Буфер для электрофореза Электрофорез проводится в камере,
- 25. Перед началом электрофореза к образцам добавляют два
- 26. В образец добавляется глицерин(Loading buffer) + краситель(loading
- 27. EtBr (бромистый этидий) является наиболее распространенным реагентом,
- 28. Картирование и секвенирование - наиболее важные и
- 29. Карты генетического сцепления строят, наблюдая за тем,
- 30. Хромосомные карты показывают расположение генов или отдельных
- 31. Карта X-хромосомы человека
- 32. Графическое представление нормального человеческого кариотипа в виде идеограмм всех его хромосом
- 33. Последняя стадия физического картирования генома
- 34. Методы секвенирования, то есть прочтения последовательности нуклеотидов
- 35. Techniques of nucleic acid sedimentation. (a)
- 37. Метод Сэнгера (плюс-минус метод) — метод секвенирования(определения последовательности нуклеотидов) ДНК, также
- 38. Метод включал два этапа. Сначала в ограниченных
- 39. Секвенирование ДНК по Максаму-Гилберту (метод терминаторов) -
- 42. ДНК-секвенирование
- 43. Геномные анализы помогают выявить Хромосомные болезни — наследственные
- 44. Болезни, обусловленные нарушением числа хромосом синдром Дауна — трисомия по
- 45. Благодаря геномным исследованиям, а также совершенствованию инструментальной
- 46. Геномные исследования и идентификация генов, повреждение которых
- 47. Результаты геномных исследований все шире используются в
- 49. ПЦР
- 50. Один из ранних методов, основанный на использовании

Слайд 2ОБЩАЯ СТРАТЕГИЯ ИССЛЕДОВАНИЯ МАКРОМОЛЕКУЛ
Получение ОБЪЕКТА исследования
клетки тканей растений и животных)
Обработка
Выделение нужной фракции или нужной макроструктуры (мембраны, органеллы, ядро, рибосомы, микросомы, цитоплазмы и др.)
Обработка препарата. Выделение фракции белков (липидов,
ДНК, РНК и т.д.)
Детекция
нужной молекулы
Выделение и очистка
нужной молекулы
1
2
3
4
5
6
Э
К
С
Т
Р
А
К
Ц
И
Я
Обработка объекта исследования (лизис, разрушение,")
Слайд 3Геномный анализ
Геномный анализ — это один из цитогенетических методов исследования, изучающий число геномов
при диагностике наследственных болезней.


Слайд 5Выделение и очистка ДНК
Основная цель этапов выделения ДНК - последовательная очистка
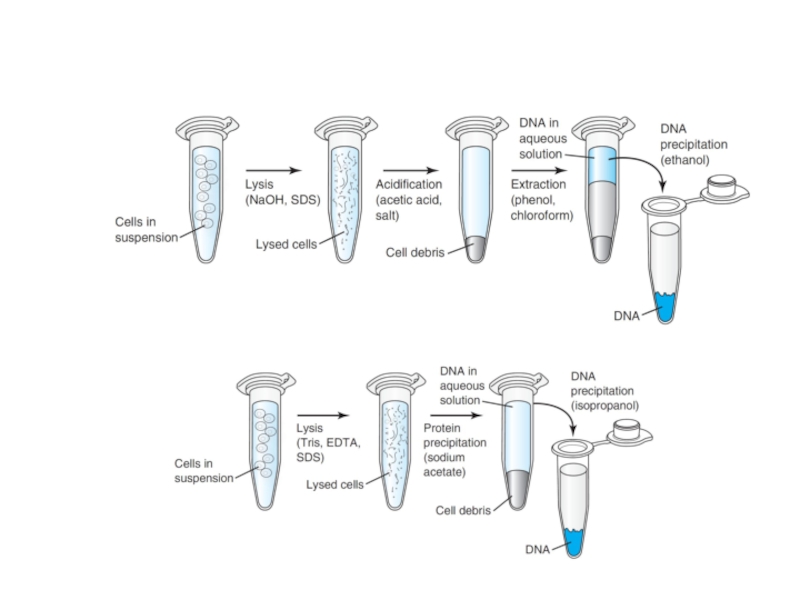
Этапы: 1. Гомогенизация ткани и изолирование ядерной ДНК. Экстрагирование ДНК из ядра проводят с помощью буферного солевого раствора, содержащего детергент SDS, который способствует лизированию ядра. Детергент также ингибирует активность нуклеаз во время экстрагирования.
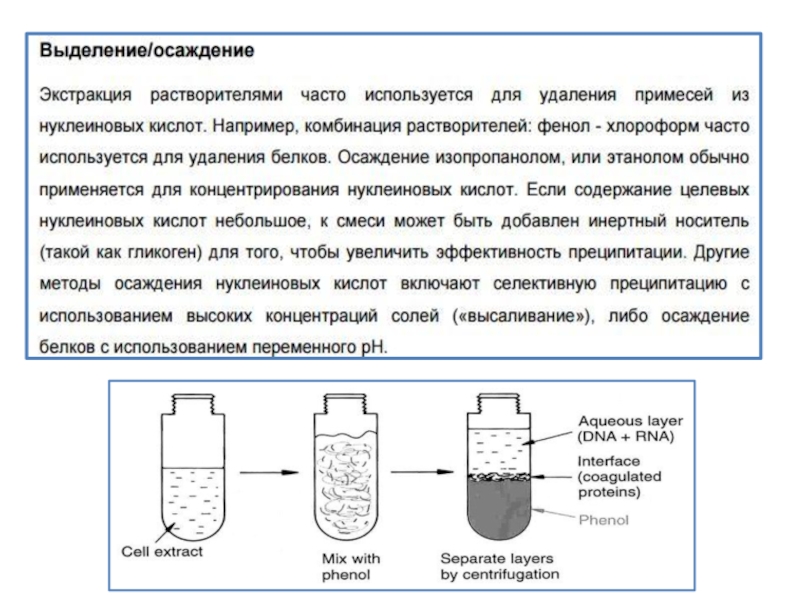
2. Депротеинизация фенолом или смесью Фенол:хлороформ. Фенол активизирует денатурацию белков вследствии утраты солюбилитивной способности (растворяемости) и выпадения в осадок (преципитат) из раствора, содержащего фенол+буферный солевой раствор. (Смесь перемешивают и центрифугируют для отделения осажденных белков от фракции НК. Надосадочную жидкость отбирают и повторяют цикл перемешивания с фенолом и центрифугированием до полного осаждения белков)
Преципитация НК охлажденным этанолом . Холодный этанол приводит к расслоению жидкого раствора (ДНК будет содержаться в верхней части раствора НК-т). Фракцию ДНК отбирают на границе раздела фаз между этанолом и физраствором . РНК остается на дне микропробирки.
4. Редиссольвация (растворение) ДНК и обработка рибонуклеазой для удаления контаминирующей РНК. (для полной очистки от остатков белков также можно использовать протеазу).
При выделении РНК на финальном этапе вместо рибонуклеазы используют ДНазу (дезоксирибонуклеазу) . В более упрощенной схеме выделения РНК – ткань гомогенизируют в растворе, содержащем 4М гуанидин тиоционат. РНК экстракт смешивают с фенолом и хлороформом (или бром-хлор-пропаном) при встряхивании, затем смесь центрифугируют . РНК отбирают из надосадочной жидкости (ДНК и белки находятся на границе раздела двух фаз)








Слайд 15Существует несколько методов ДНК–тестирования (анализа)
Электрофорез ДНК в агарозном геле, в полиакриламидном
Анализ полиморфизма длины рестрикционных фрагментов — ПДРФ
Метод полимеразной цепной реакции — ПЦР
Секвенирование ДНК
Детекция ДНК на чипах
Электрофорез ДНК в агарозном геле, в полиакриламидном гелеАнализ полиморфизма длины рестрикционных")
Слайд 16Электрофорез – метод разделения макромолекул, различающихся по таким параметрам, как размеры
, пространственная")

Слайд 18Преимущества агарозного геля
прочность агарозного геля;
крупнопористость (позволяющая разделять особенно крупные молекулы, в
агарозный гель является очень мягким носителем (т.е. в отличие от электрофореза на бумаге, например, при нем не происходит инактивации белков, что позволяет определять активность отдельных фракций белков после проведения электрофореза);
приготовление агарозного геля значительно проще, чем крахмального и полиакриламидного;
относительно небольшая продолжительность электрофореза (сильно варьирует в зависимости от варианта метода);
относительная дешевизна метода.
;агарозный гель")
Слайд 19Агароза очень хрупка, легко разрушается при манипулировании. Агарозные гели имеют «поры»
Разделение в агарозных гелях происходит быстро, но с ограниченным разрешением, так как полосы, образующиеся в агарозных гелях, имеют тенденцию размываться и распространяться в стороны. Это является результатом большого размера пор и не может быть предотвращено. Агарозные гели получают суспендированием сухого порошка агарозы в водном буфере, и кипячением смеси до того момента, когда агароза расплавится и образует прозрачный раствор. Затем раствор наливают на подложку и дают остыть до комнатной температуры, чтобы сформировался прочный гель. При застывании агароза формирует матрикс, плотность которого определяется концентрацией.

Слайд 20При проведении электрофореза фрагменты ДНК мигрирют в геле под воздействием сил

Слайд 21Разделение фрагментов ДНК происходит из-за наличия у них заряда. Фосфатные остатки
Фрагменты ДНК, имеющие наименьшую длину, приближаются быстрее всего к + электроду, в то время как длинные фрагменты остаются максимально близко к минусу. Те же базовые принципы электрофореза могут использоваться для разделения РНК и протеинов. Увеличение концентрации агарозы в геле уменьшает скорость миграции ДНК и позволяет разделять малые фрагменты ДНК. Чем больше напряжение тем быстрее проходит форез - но слишком сильное напряжение нагреет буфер а это недопустимо. Конформация ДНК тоже играет важную роль. Кольцевые плазмиды(неразрезанные рестриктазами) двигаются с другой скоростью чем линейные или суперскрученные.


Слайд 23В зависимости от выбранной процентности агарозы статистический размер ячеек геля будет

Слайд 24Буфер для электрофореза
Электрофорез проводится в камере, заполненной буферным раствором. Чаще всего

Слайд 25Перед началом электрофореза к образцам добавляют два разных красителя с кислым

Слайд 26В образец добавляется глицерин(Loading buffer) + краситель(loading dye) концентрация молекул ДНК
 + краситель(loading dye) концентрация молекул ДНК в образце желательно >")
Слайд 27EtBr (бромистый этидий) является наиболее распространенным реагентом, который используется для окрашивания
 является наиболее распространенным реагентом, который используется для окрашивания ДНК в агарозном геле.")
Слайд 28Картирование и секвенирование - наиболее важные и трудоемкие части всех геномных
Картирование - определение положения фрагментов ДНК в хромосоме с использованием генетических методов, то есть анализ сцепления и рекомбинации генов на основе родословных. Карты, полученные при использовании данного метода, часто называют картами генетического сцепления.
Маркеры, используемые в картировании, должны быть полиморфными, то есть должны существовать альтернативные формы признака, которые можно было бы легко отличить и описать у членов семьи.

Слайд 29Карты генетического сцепления строят, наблюдая за тем, как часто два маркера
Два маркера, расположенные на одной хромосоме вблизи друг друга, имеют тенденцию передаваться от родителей ребенку совместно. Во время нормальных процессов формирования сперматозоидов и яйцеклеток цепочка ДНК случайным образом разрывается и воссоединяется в тех или иных местах хромосомы или ее копии (то есть гомологичной хромосомы).
Этот процесс, называемый мейотической рекомбинацией, может приводить к разделению двух маркеров, первоначально расположенных на одной хромосоме. Чем ближе расположены маркеры друг к другу, тем "теснее" они сцеплены, тем менее вероятно, что процесс рекомбинации разделит их.

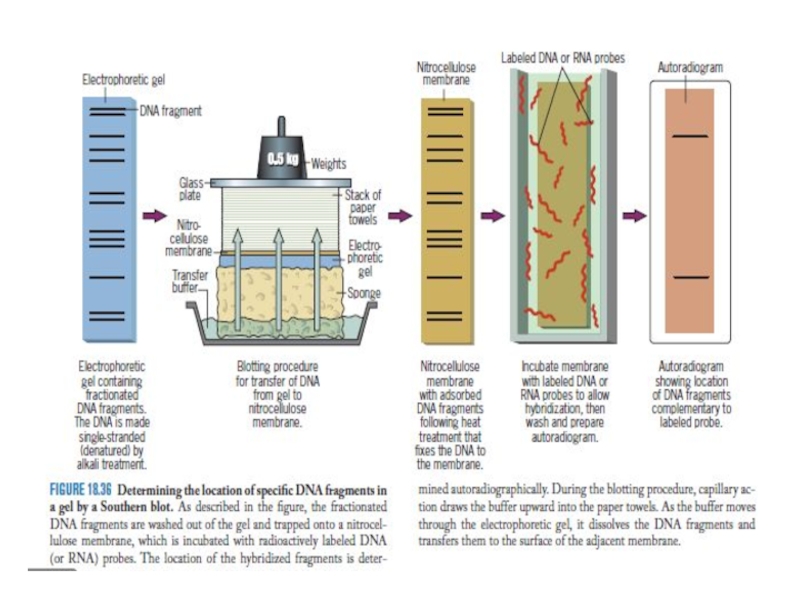
Слайд 30Хромосомные карты показывают расположение генов или отдельных участков ДНК на соответствующих
Эти маркеры могут быть физически связаны с определенным сегментом хромосомы при гибридизации in situ - методе, который позволяет пометить ДНК специальной "видимой" (флуоресцентной или радиоактивной) меткой. Локализация меченого зонда устанавливается после того, как он свяжется с комплементарной цепочкой ДНК на интактной хромосоме.


Слайд 32Графическое представление нормального человеческого
кариотипа в виде идеограмм
всех его хромосом

Слайд 33
Последняя стадия физического картирования генома человека - определение всех пар оснований

Слайд 34Методы секвенирования, то есть прочтения последовательности нуклеотидов в молекуле ДНК, дают

Слайд 35Techniques of nucleic acid sedimentation.
(a) Separation of different-sized DNA molecules
(b) Separation of DNA molecules by equilibrium sedimentation on the basis of differences in base composition. The DNA sample is mixed with the CsCl solution (step 1) and subjected to extended centrifugation (e.g., 50,000 rpm for 72 hours). The CsCl gradient forms during the centrifugation (step 2), and the DNA molecules band in regions of equivalent density (step 3). (c) The tube from the experiment of b is punctured and the contents are allowed to drip into successive tubes, thereby fractionating the tube’s contents. The absorbance of the solution in each fraction is measured and plotted as shown.
 Separation of different-sized DNA molecules by velocity sedimentation. The")
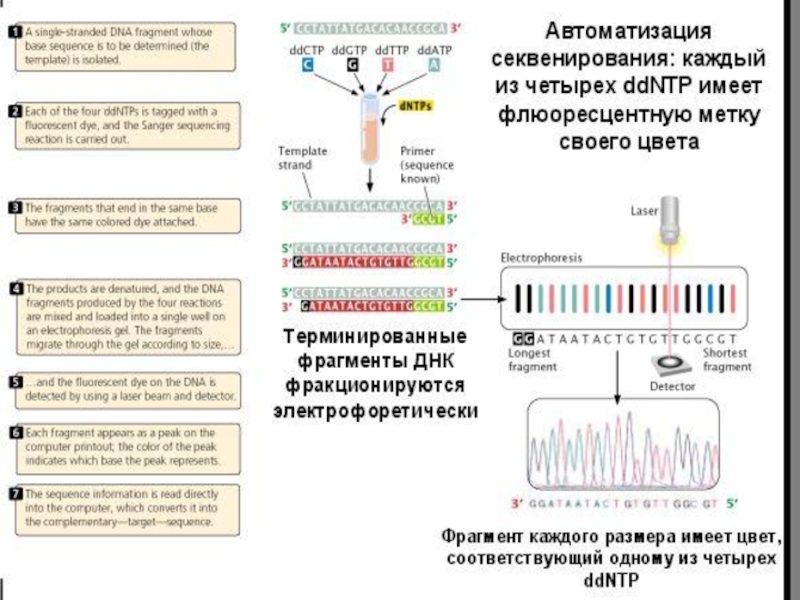
Слайд 37Метод Сэнгера (плюс-минус метод) — метод секвенирования(определения последовательности нуклеотидов) ДНК, также известен как метод обрыва
Впервые этот метод секвенирования был предложен Фредериком Сэнгером в 1977 году, за что он был удостоен Нобелевской премии по химии в 1980 году.
Принцип метода
В классическом варианте метода Сэнгера одна из цепочек анализируемой ДНК выступает в качестве матрицы для синтеза комплементарной цепочки ферментом ДНК-полимеразой, в качестве праймеров - синтетические олигонуклеотиды или природные субфрагменты, получаемые при гидролизе рестрицирующими эндонуклеазами.
 — метод секвенирования(определения последовательности нуклеотидов) ДНК, также известен как метод обрыва цепи или методом прямого")
Слайд 38Метод включал два этапа. Сначала в ограниченных условиях проводили полимеразную реакцию
В результате, в "минус" системе терминация происходила перед dNTP данного типа, а в "плюс" системе - после него.
Полученные таким образом восемь образцов разделяли с помощью электрофореза, "считывали" сигнал и определяли последовательность исходной ДНК. Этим способом была секвенирована короткая ДНК фага фХ174, состоящая из 5386 нуклеотидных пар.

Слайд 39Секвенирование ДНК по Максаму-Гилберту (метод терминаторов) - Этот метод также называется
Центральный элемент метода секвенирования ДНК по Максаму-Гилберту - химическая деградация меченой цепи ДНК изменился за эти годы не так сильно. Хотя многими исследователями и предлагались различные варианты модификаций тех или иных азотистых оснований, все же в основном арсенале этого метода находятся 4-6 химических реакций, показывающих наиболее стабильные результаты. К некоторому преимуществу метода секвенирования ДНК химической деградацией можно отнести то, что здесь определяется последовательность фрагмента ДНК, или геномного, или клонированного, в каком-либо подходящем векторе (т.е. реплицировавшегося in vivo), а не новосинтезированная in vitro копия, как в ферментативном методе с дидезокситерминаторами. Еще одно отличие метода секвенирования ДНК по Максаму-Гилберту от метода Сэнгера заключается в том, что его осуществление может начаться практически с любого сайта узнавания какой-нибудь рестрикционной эндонуклеазы, присутствующего во вставке и поэтому не требуется предварительного знания даже небольшого участка нуклеотидной последовательности, окружающего данное место.
 - Этот метод также называется секвенированием ДНК методом химической")


Слайд 43Геномные анализы помогают выявить
Хромосомные болезни — наследственные заболевания, обусловленные изменением числа или
К хромосомным относятся болезни, обусловленные геномными мутациями или структурными изменениями отдельных хромосом. Хромосомные болезни возникают в результате мутаций в половых клетках одного из родителей.

Слайд 44Болезни, обусловленные нарушением числа хромосом
синдром Дауна — трисомия по 21-й хромосоме,
синдром Патау — трисомия по
синдром Эдвардса — трисомия по 18-й хромосоме
Болезни, связанные с нарушением числа половых хромосом
Синдром Шерешевского — Тёрнера — отсутствие одной Х-хромосомы у женщин (45 ХО) вследствие нарушения расхождения половых хромосом
полисомия по Х-хромосоме — включает трисомию (кариотии 47, XXX), тетрасомию (48, ХХХХ), пентасомию (49, ХХХХХ), отмечается незначительное снижениеинтеллекта, повышенная вероятность развития психозов и шизофрении с неблагоприятным типом течения;
полисомия по Y-хромосоме — как и полисомия по X-хромосоме, включает трисомию (кариотии 47, XYY), тетрасомию (48, ХYYY), пентасомию (49, ХYYYY), клинические проявления также схожи с полисомией X-хромосомы;

Слайд 45Благодаря геномным исследованиям, а также совершенствованию инструментальной диагностической медицинской техники появилась
Более того, возможна преимплантационная диагностика, когда ряд яйцеклеток матери оплодотворяются in vitro (в пробирке), затем несколько зародышей развиваются до стадии 8 клеток, и 1 - 2 клетки зародыша анализируют на наличие поврежденного гена. Зародыш, не содержащий поврежденного гена, имплантируется в клетку.

Слайд 46Геномные исследования и идентификация генов, повреждение которых приводит к заболеваниям, позволяет

Слайд 47Результаты геномных исследований все шире используются в судебной медицине.
Появление технологии
")

Слайд 50Один из ранних методов, основанный на использовании особых ферментов — рестриктаз.
Анализ полиморфизма длины рестрикционных фрагментов - ПДРФ