- Главная
- Разное
- Дизайн
- Бизнес и предпринимательство
- Аналитика
- Образование
- Развлечения
- Красота и здоровье
- Финансы
- Государство
- Путешествия
- Спорт
- Недвижимость
- Армия
- Графика
- Культурология
- Еда и кулинария
- Лингвистика
- Английский язык
- Астрономия
- Алгебра
- Биология
- География
- Детские презентации
- Информатика
- История
- Литература
- Маркетинг
- Математика
- Медицина
- Менеджмент
- Музыка
- МХК
- Немецкий язык
- ОБЖ
- Обществознание
- Окружающий мир
- Педагогика
- Русский язык
- Технология
- Физика
- Философия
- Химия
- Шаблоны, картинки для презентаций
- Экология
- Экономика
- Юриспруденция
Генные заболевания презентация
Содержание
- 1. Генные заболевания
- 2. Генные заболевания (ГЗ) это разнородная по клиническим
- 3. Этиология (генные мутации, которые обусловливают наследственные заболевания)
- 4. Механизмы развития ГЗ (варианты первичных мутантных аллелей)
- 5. Варианты нарушений функций белка при ГЗ Потеря
- 6. Фенотипические эффекты ГЗ (клинические) Дизморфогенез (врожденные пороки развития) Нарушенный обмен веществ Смешанные эффекты
- 7. Сроки реализации патологических мутаций Внутриутробно – 25%
- 8. Генокопии – заболевания со сходной клиникой,
- 9. Классификация генных заболеваний По генетическому принципу:
- 10. По клиническому принципу (продолжение): Нервные Нервно-мышечные
- 11. Патогенетический принцип (продолжение): Нарушение обмена веществ (углеводный,
- 12. Общие закономерности патогенеза Мутантный аллель Патологический первичный
- 13. Ахондроплазия. Частота встречаемости – 1:100 000 Соотношение
- 15. Ахондроплазия (продолжение)
- 16. Синдром Марфана Частота встречаемости – 1:10 000
- 17. Синдром Марфана Характерен клинический полиморфизм: Костно-мышечная система
- 18. Синдром Марфана
- 19. Синдром Элерса-Данлоса Гетерогенное наследственное заболевание соединительной ткани
- 20. Критерии диагностики: Кожа – гиперрастяжимость, бархатистость, кровоточивость,
- 21. Синдром Эллерса-Данлоса
- 22. Адреногенитальный синдром (врожденная гиперплазия коры надпочечников) Популяционная
- 23. Адреногенитальный синдром Сольтеряющая – полный дефицит фермента, проявляется
- 24. Простая вирильная форма На фото представлена 42-летняя
- 25. Нарушение аминокислотного обмена - фенилкетонурия Частота встречаемости
- 26. Скрининг-тесты: выявление фенилпировиноградной кислоты
- 27. Диагностика дети рождаются здоровыми,
- 28. Лечение безбелковая диета с гидролизатами белка, которые
- 29. Целиакия. (глютеновая болезнь, болезнь Ги-Гертера-Гейбнера) – непереносимость
- 31. Врожденный гипотиреоз Частота встречаемости – 1:3500 –
- 32. Муковисцидоз (особенности наследования) Отец носитель ребенок
- 33. Патогенез обусловлен мутациями в гене CFTR, который
- 34. Форма муковисцидоза Смешанная в 75-80% Легочная 15-20%
- 35. Поражение органов дыхания У 1/3 больных М
- 36. Поражение ЖКТ Мекониальный илеус (период новорожденности) Гемолитическая
- 37. Осложнение - бесплодие Случаи полного бесплодия у
- 39. Генетический анализ (медико-генетическое консультирование) Пренатальная диагностика
- 40. Потовый тест Нужно не менее 100мг потовой
- 41. Диета Основной принцип – повышение энергетической ценности
- 42. Заместительная терапия - ферменты Должна быть пожизненной,
- 43. Антибактериальная терапия До 2 лет жизни ребенка
- 44. Ацетилцистеин (ингаляци, сироп) Амбробене (ингаляци, сироп) Рекомбинантная
- 45. Спасибо за внимание!

Слайд 2Генные заболевания (ГЗ)
это разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями
на генном уровне
Формы генных мутаций:
Полные обусловлены мутациями в гаметах. В этом случае патологические гены обнаруживают во всех клетках организма
Мозаичные обусловлены мутациями, кот.возникают на ранних стадиях дробления зиготы в одной из клеток
Формы генных мутаций:
Полные обусловлены мутациями в гаметах. В этом случае патологические гены обнаруживают во всех клетках организма
Мозаичные обусловлены мутациями, кот.возникают на ранних стадиях дробления зиготы в одной из клеток
это разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями на генном уровнеФормы генных")
Слайд 3Этиология (генные мутации, которые обусловливают наследственные заболевания)
Точечные мутации – замена одного
нуклеотида другим комплементарным или некомплементарным
Миссенс мутации – замена нуклеотида, который сопровождается заменой аминокислотного шифра кодона и ведет к замене аминокислоты в составе белка
Нонсенс – замена нуклеотида, который приводит к замещению информационно значимого кодона сто-кодоном и сопровождается преждевременным обрывом трансляции
Делеции – мутация, который ведет к потере определенного участка молекулы ДНК
Инсерции – вставки определенного участка молекулы ДНК в структуру гена
Сдвиг рамки считывания – вследствие вставки, дупликации или делеции
Нарушение сплайсинга - нарушение созревания РНК
Экспансия (увеличение числа) тринуклеотидных повторов
Миссенс мутации – замена нуклеотида, который сопровождается заменой аминокислотного шифра кодона и ведет к замене аминокислоты в составе белка
Нонсенс – замена нуклеотида, который приводит к замещению информационно значимого кодона сто-кодоном и сопровождается преждевременным обрывом трансляции
Делеции – мутация, который ведет к потере определенного участка молекулы ДНК
Инсерции – вставки определенного участка молекулы ДНК в структуру гена
Сдвиг рамки считывания – вследствие вставки, дупликации или делеции
Нарушение сплайсинга - нарушение созревания РНК
Экспансия (увеличение числа) тринуклеотидных повторов
Точечные мутации – замена одного нуклеотида другим комплементарным или")
Слайд 4Механизмы развития ГЗ
(варианты первичных мутантных аллелей)
Отсутствие синтеза полипептидной цепи (белка)
Синтез
аномальной по первичной структуре полипептидной цепи
Количественно недостаточный синтез полипептидной цепи
Количественно избыточный синтез полипептидной цепи
Количественно недостаточный синтез полипептидной цепи
Количественно избыточный синтез полипептидной цепи
 Отсутствие синтеза полипептидной цепи (белка)Синтез аномальной по первичной")
Слайд 5Варианты нарушений функций белка при ГЗ
Потеря функции – за счет ингибирования
процесса транскрипции/трансляции или за счет изменения функциональных свойств
Появление новой функции - у мутантного белка вместе с нормальной функцией появляются новые цитотоксические свойства, которые приводят к гибели клеток
Доминантный негативный эффект – первичный продукт мутантного аллеля ингибирует функцию нормальных белков
Изменение дозы гена (делеции или дупликации) – может приводить к нарушению пространственной конфигурации молекулярного продукта
Появление новой функции - у мутантного белка вместе с нормальной функцией появляются новые цитотоксические свойства, которые приводят к гибели клеток
Доминантный негативный эффект – первичный продукт мутантного аллеля ингибирует функцию нормальных белков
Изменение дозы гена (делеции или дупликации) – может приводить к нарушению пространственной конфигурации молекулярного продукта

Слайд 6Фенотипические эффекты ГЗ (клинические)
Дизморфогенез (врожденные пороки развития)
Нарушенный обмен веществ
Смешанные эффекты
Дизморфогенез (врожденные пороки развития)Нарушенный обмен веществСмешанные эффекты")
Слайд 7Сроки реализации патологических мутаций
Внутриутробно – 25%
В допубертатном периоде – 45%
Пубертат, юношеский
возраст – 20%
Старше 20 лет – 10%
Старше 20 лет – 10%

Слайд 8
Генокопии – заболевания со сходной клиникой, но различной генетической основой (мутации
в разных локусах)
Фенокопии – фенотипические проявления, имитирующие ГЗ (в случае действия внешних факторов внутриутробно)
Нормокопирование – в случае, когда мутантный генотип не проявляется фенотипически (при медикаментозном воздействии, либо коррекции диетотерапией и т.д.)
Фенокопии – фенотипические проявления, имитирующие ГЗ (в случае действия внешних факторов внутриутробно)
Нормокопирование – в случае, когда мутантный генотип не проявляется фенотипически (при медикаментозном воздействии, либо коррекции диетотерапией и т.д.)
Фенокопии –")
Слайд 9Классификация генных заболеваний
По генетическому принципу:
Аутосомно-доминантные
Аутосомно-рецессивны
Х-сцепленные доминантные
Х-сцепленные рецессивные
У-сцепленные (голандрические)
Митохондриальные
Митохондриальные")
Слайд 10По клиническому принципу (продолжение):
Нервные
Нервно-мышечные
Кожные
Глазные
Болезни опорно-двигательного аппарата и др. (в зависимости от
системы, наиболее вовлеченной в патпроцесс)
:НервныеНервно-мышечныеКожныеГлазныеБолезни опорно-двигательного аппарата и др. (в зависимости от системы, наиболее вовлеченной в патпроцесс)")
Слайд 11Патогенетический принцип (продолжение):
Нарушение обмена веществ (углеводный, аминокислотный, обмен витаминов, липидов, металлов
и др.)
Врожденные пороки развития
Комбинированные состояния
Врожденные пороки развития
Комбинированные состояния
:Нарушение обмена веществ (углеводный, аминокислотный, обмен витаминов, липидов, металлов и др.)Врожденные пороки развитияКомбинированные состояния")
Слайд 12Общие закономерности патогенеза
Мутантный аллель
Патологический первичный продукт
Цепь последующих биохимических процессов
Клетки
Органы
Организм

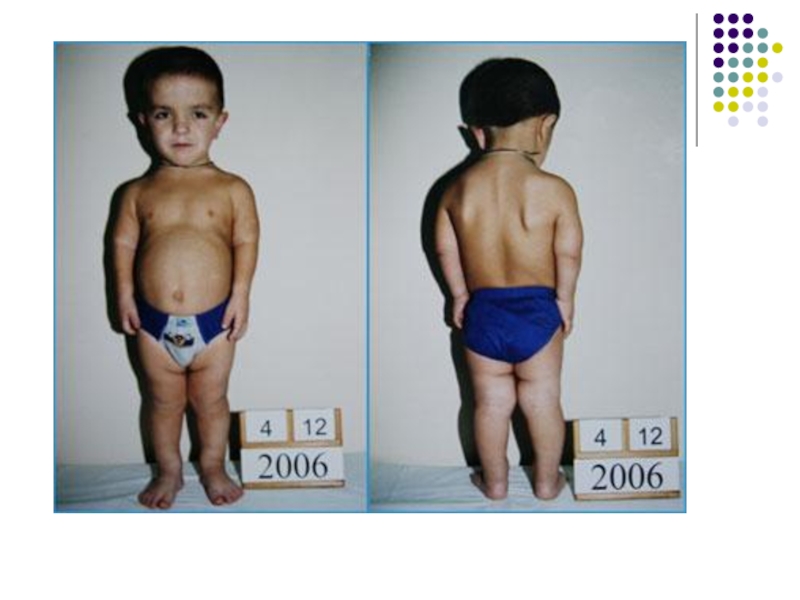
Слайд 13Ахондроплазия.
Частота встречаемости – 1:100 000
Соотношение полов – 1:1
Критерии диагностики:
Низкий рост
Большой
череп с выступающим затылком
Вогнутая переносица
Прогнатизм у взрослых
Укорочение конечностей за счет укорочения проксимальных отделов
Кисти широкие, короткие, пальцы в виде тризубца вараженный поясничный лордоз
Интеллект не страдает
Диагностика портретная
Медико-генетическое консультирование: риск для потомков 50%. Пренатальная диагностика в 24-26 недель беременности
Лечение симптоматическое. Ортопедическая коррекция.
Вогнутая переносица
Прогнатизм у взрослых
Укорочение конечностей за счет укорочения проксимальных отделов
Кисти широкие, короткие, пальцы в виде тризубца вараженный поясничный лордоз
Интеллект не страдает
Диагностика портретная
Медико-генетическое консультирование: риск для потомков 50%. Пренатальная диагностика в 24-26 недель беременности
Лечение симптоматическое. Ортопедическая коррекция.

")
Слайд 16Синдром Марфана
Частота встречаемости – 1:10 000
Соотношение полов – 1:1
75% - больны
родители; 25% - результат новой мутации
Доминантное заболевание соединительной ткани
Причина – мутации в гене фибриллина, который отвечает за синтез соединительнотканного белка фибриллина и как следствие – повышенная растяжимость соединительной ткани
Доминантное заболевание соединительной ткани
Причина – мутации в гене фибриллина, который отвечает за синтез соединительнотканного белка фибриллина и как следствие – повышенная растяжимость соединительной ткани

Слайд 17Синдром Марфана
Характерен клинический полиморфизм:
Костно-мышечная система – арахнодактилия, высокий рост, деформации грудной
клетки, гипермобильность суставов, плоскостопие, мышечная гипотония, недоразвитие вертлужной впадины и др.
Глаза – подвывих хрусталика, миопия, отслойка сетчатки, уплощение роговицы, увеличение длины оси глазного яблока
ССС – аортальная регургитация, расслоение аорты, митральная регургитация, застойные сердечные нарушения, дизритмия и др.
Респираторный тракт – спонтанный пневмоторакс
ЦНС – эктазия твердой мозговой оболочки, аномалии развития
Кожа – паховые грыжи, атрофические стрии
Диагностика: повышение в моче уровня оксипролина и глюкозаминогликанов
Глаза – подвывих хрусталика, миопия, отслойка сетчатки, уплощение роговицы, увеличение длины оси глазного яблока
ССС – аортальная регургитация, расслоение аорты, митральная регургитация, застойные сердечные нарушения, дизритмия и др.
Респираторный тракт – спонтанный пневмоторакс
ЦНС – эктазия твердой мозговой оболочки, аномалии развития
Кожа – паховые грыжи, атрофические стрии
Диагностика: повышение в моче уровня оксипролина и глюкозаминогликанов


Слайд 19Синдром Элерса-Данлоса
Гетерогенное наследственное заболевание соединительной ткани с различными типами наследования
АД тип
– 1,7,8
АР тип – 6
Х-сцепленный – 5 и 9 типы
Всего 10 типов СЭД
АР тип – 6
Х-сцепленный – 5 и 9 типы
Всего 10 типов СЭД

Слайд 20Критерии диагностики:
Кожа – гиперрастяжимость, бархатистость, кровоточивость, стрии в области поясницы, рубцы,
подкожные узелки
Конечности – варикозные вены, подкожные узелки на голенях
Суставы – пассивное разгибание в суставах кистей, переразгибание крупных суставов (в зависимости от типа СЭД)
Глаза – птоз, отслойка сетчатки, разрыв глазного яблока
Зубы – частичная адонтия, опалесцирующая эмаль, пародонтоз, множественный кариес
Уши – гиперрастяжимость
Грудная клетка – сколиоз, кифоз, лордоз, плоская спина, вдавление грудины
Сердце – ПМК, аритмии, ВСД
Живот – грыжи, спонтанная перфорация кишечника
Внутренние органы – птоз желудка, почек и матки
Мозг – аневризма сосудов, субарахноидальные кровоизлияния
Стремительные роды
Конечности – варикозные вены, подкожные узелки на голенях
Суставы – пассивное разгибание в суставах кистей, переразгибание крупных суставов (в зависимости от типа СЭД)
Глаза – птоз, отслойка сетчатки, разрыв глазного яблока
Зубы – частичная адонтия, опалесцирующая эмаль, пародонтоз, множественный кариес
Уши – гиперрастяжимость
Грудная клетка – сколиоз, кифоз, лордоз, плоская спина, вдавление грудины
Сердце – ПМК, аритмии, ВСД
Живот – грыжи, спонтанная перфорация кишечника
Внутренние органы – птоз желудка, почек и матки
Мозг – аневризма сосудов, субарахноидальные кровоизлияния
Стремительные роды


Слайд 22Адреногенитальный синдром (врожденная гиперплазия коры надпочечников)
Популяционная частота – 1:5000 новорожденных
Аутосомно-рецессивное заболевание
Относится
к группе наследственных нарушений биосинтеза стероидных гормонов. Известно 5 разновидностей наследственных дефицитов ферментов
Наиболее распространенная форма – дефицит 21-гидроксилазы, имеющая 4 клинических варианта:
Сольтеряющая
Простая вирильная
Поздняя (неклассическая)
Латентная (бессимптомная)
Наиболее распространенная форма – дефицит 21-гидроксилазы, имеющая 4 клинических варианта:
Сольтеряющая
Простая вирильная
Поздняя (неклассическая)
Латентная (бессимптомная)
Популяционная частота – 1:5000 новорожденныхАутосомно-рецессивное заболеваниеОтносится к группе наследственных нарушений")
Слайд 23Адреногенитальный синдром
Сольтеряющая – полный дефицит фермента, проявляется нарушением солевого обмена (дефицит минералокортикоидов).
С первых дней – срыгивания, рвота, недостаточность периферического кровообращения, сонливость. Биохимия – гиперкалиемия, гипонатриемия, ацидоз.
Простая вирильная – прогрессирующая вирилдизация, раннее соматическое развитие. У девочек – разная степень маскулинизации. Диагноз ставитсмя на 5-7 году жизни при появлении признаков преждевременного полового развития.
Поздняя (неклассическая вирилизующая) форма – проявляется в подростковом возрасте: симптомы избытка андрогенов у девочек – ускорение костного роста, нарушение менструального цикла, гирсутизм; у мальчиков – ускорение костного роста, преждевременное оволосение
Латентная форма – нет клинических проявлений, но в сыворотке – повышение уровня предшественников кортизола
Простая вирильная – прогрессирующая вирилдизация, раннее соматическое развитие. У девочек – разная степень маскулинизации. Диагноз ставитсмя на 5-7 году жизни при появлении признаков преждевременного полового развития.
Поздняя (неклассическая вирилизующая) форма – проявляется в подростковом возрасте: симптомы избытка андрогенов у девочек – ускорение костного роста, нарушение менструального цикла, гирсутизм; у мальчиков – ускорение костного роста, преждевременное оволосение
Латентная форма – нет клинических проявлений, но в сыворотке – повышение уровня предшественников кортизола
. С первых дней –")
Слайд 24Простая вирильная форма
На фото представлена 42-летняя низкорослая (146 см) пациентка с
адреногенитальным синдромом (АГС) с мужской внешностью, низким голосом, выраженной мускулатурой, широкой грудной клеткой, неполной лысиной и мужским типом оволосения (гирсутизм).
У пациентки в возрасте 6 лет возникли признаки преждевременного полового созревания, в 8 лет прекратился рост (преждевременное закрытие эпифизов). Причиной АГС в данном случае является дефицит 21-гидроксилазы.
У пациентки в возрасте 6 лет возникли признаки преждевременного полового созревания, в 8 лет прекратился рост (преждевременное закрытие эпифизов). Причиной АГС в данном случае является дефицит 21-гидроксилазы.
 пациентка с адреногенитальным синдромом (АГС) с")
Слайд 25Нарушение аминокислотного обмена - фенилкетонурия
Частота встречаемости – 1:10 000, частота гетерозиготного
носительства – 1:50 – 1:100
Соотношение полов – 1:1
Тип наследования – аутосомно-рецессивный, тяжесть течения зависит от экспрессивности гена
Патогенез – недостаточность фенилаланингидроксилазы, в результате чего ФА не превращается в тирозин. Продукты метаболизма влияют на развитие нервной системы (нарушается формирование миелиновой оболочки, синтез ГАМК и серотонина). Нарушение синтеза тирозина ведет к снижению синтеза меланина, адреналина, тироксина, дофамина
Возможна молекулярно-генетическая пренатальная диагностика и выявление гетерозигот
Соотношение полов – 1:1
Тип наследования – аутосомно-рецессивный, тяжесть течения зависит от экспрессивности гена
Патогенез – недостаточность фенилаланингидроксилазы, в результате чего ФА не превращается в тирозин. Продукты метаболизма влияют на развитие нервной системы (нарушается формирование миелиновой оболочки, синтез ГАМК и серотонина). Нарушение синтеза тирозина ведет к снижению синтеза меланина, адреналина, тироксина, дофамина
Возможна молекулярно-генетическая пренатальная диагностика и выявление гетерозигот

Слайд 26
Скрининг-тесты:
выявление фенилпировиноградной кислоты в моче или плазме в роддоме;
с трихлорным железом)
")
Слайд 27
Диагностика
дети рождаются здоровыми, но с 2-х месяцев (при поступлении
с молоком матери ФА) развивается клиника:
дерматит
осветление волос и радужки,
генерализованные судороги
умственная отсталость
микроцефалия «мышиный» запах тела,
гиперрефлексия
повышенный тонус мышц
дерматит
осветление волос и радужки,
генерализованные судороги
умственная отсталость
микроцефалия «мышиный» запах тела,
гиперрефлексия
повышенный тонус мышц
 развивается")
Слайд 28Лечение
безбелковая диета с гидролизатами белка, которые не содержат ФА до 19-ти
лет под контролем ФА (в пределах 3,5 мг%)

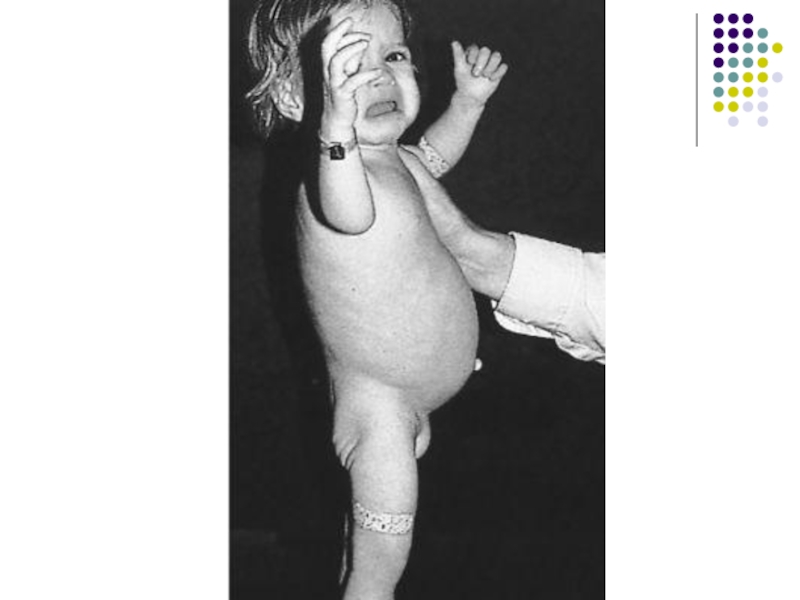
Слайд 29Целиакия.
(глютеновая болезнь, болезнь Ги-Гертера-Гейбнера) – непереносимость глиадина, который содержится в клейковине
Частота
встречаемости – 1:3000
Аутосомно-доминантный тип с неполной пенетрантностью
Клиника появляется после введения продуктов, которые содержат глютен (хлеб, манная каша)
Постоянная стеаторея, потеря массы тела, дефицитные состояния
Лечение – безглютеновая диета (рисовая, соевая, гречаная мука, обезжиренное молоко)
Аутосомно-доминантный тип с неполной пенетрантностью
Клиника появляется после введения продуктов, которые содержат глютен (хлеб, манная каша)
Постоянная стеаторея, потеря массы тела, дефицитные состояния
Лечение – безглютеновая диета (рисовая, соевая, гречаная мука, обезжиренное молоко)
 – непереносимость глиадина, который содержится в клейковинеЧастота встречаемости – 1:3000Аутосомно-доминантный тип")
Слайд 31Врожденный гипотиреоз
Частота встречаемости – 1:3500 – 1:4000
Отношение полов – 1:1
Причины:
Дисгенезия
щитовидной железы
Аутосомно-рецессивное нарушение синтеза гормонов щитовидной железы
Дефицит ТТГ или тиреотропинрилизинггоромона
Нечувствительность ЩЖ к ТТГ
Периферическая нечувствительность к гормонам ЩЖ вследствие действия препаратов во время беременности
Дефицит йода (эндемический кретинизм)
Критерии диагностики:
Длительная желтуха
Срыгивания, запоры
Макроглоссия, микросомия, грубый голос
Увеличение живота, пупочная грыжа
Гипорефлексия, анемия, брадикардия
Повышение ТТГ и снижение уровня Т4
Массовый скрининг – на 2-5 день жизни определение ТТГ и Т4
Лечение – L-тироксин по 0,01мг/кг/сутки
Аутосомно-рецессивное нарушение синтеза гормонов щитовидной железы
Дефицит ТТГ или тиреотропинрилизинггоромона
Нечувствительность ЩЖ к ТТГ
Периферическая нечувствительность к гормонам ЩЖ вследствие действия препаратов во время беременности
Дефицит йода (эндемический кретинизм)
Критерии диагностики:
Длительная желтуха
Срыгивания, запоры
Макроглоссия, микросомия, грубый голос
Увеличение живота, пупочная грыжа
Гипорефлексия, анемия, брадикардия
Повышение ТТГ и снижение уровня Т4
Массовый скрининг – на 2-5 день жизни определение ТТГ и Т4
Лечение – L-тироксин по 0,01мг/кг/сутки

Слайд 32Муковисцидоз
(особенности наследования)
Отец
носитель
ребенок без
гена МВ
носители
больной
Мать
носитель
АР тип
Отецносительребенок безгена МВносителибольнойМатьносительАР тип")
Слайд 33Патогенез обусловлен мутациями в гене CFTR, который кодирует структуру белка -
трансмембранного регулятора проводимости. Ген CFTR находится на 7-й хромосоме (7q31).
Происходит нарушение транспорта ионов хлора и натрия через мембраны, что ведет к чрезмерному выведению хлоридов, и, как следствие, гиперсекреции густой слизи железами.
Происходит нарушение транспорта ионов хлора и натрия через мембраны, что ведет к чрезмерному выведению хлоридов, и, как следствие, гиперсекреции густой слизи железами.
Патогенез муковисцидоза

Слайд 34Форма муковисцидоза
Смешанная в 75-80%
Легочная 15-20%
Кишечная 5%
Редкие формы
Ретенционная желтуха
Изолированная электролитная (коллаптоидная)
Мекониальный илеус
у новорожденных
Дистрофическая
Отечно-анемическая
Дистрофическая
Отечно-анемическая
Фаза обострения
ремиссии
Мекониальный илеус у новорожденныхДистрофическаяОтечно-анемическаяФаза обострения")
Слайд 35Поражение органов дыхания
У 1/3 больных М манифестирует на первом году жизни
легочными инфекциями.
Симптомы:
кашель - сухой, мучительный с трудноотделяемой мокротой, часто носит пароксизмальный характер и сопровождаться рвотой (ошибочно диагностируется коклюш)
одышка
У детей старшего возраста и взрослых кашель наиболее выражен утром, после физической нагрузки и массажа
Симптомы:
кашель - сухой, мучительный с трудноотделяемой мокротой, часто носит пароксизмальный характер и сопровождаться рвотой (ошибочно диагностируется коклюш)
одышка
У детей старшего возраста и взрослых кашель наиболее выражен утром, после физической нагрузки и массажа

Слайд 36Поражение ЖКТ
Мекониальный илеус (период новорожденности)
Гемолитическая анемия (в период новорожденности)
Инвагинация
Диарея/стеаторея
Дефицит жирорастворимых
витаминов
Отеки (гипопротеинемия)
Задержка физического развития
Выпадение прямой кишки
Дистальный интестинальный обструктивный синдром
Отеки (гипопротеинемия)
Задержка физического развития
Выпадение прямой кишки
Дистальный интестинальный обструктивный синдром
Гемолитическая анемия (в период новорожденности)Инвагинация Диарея/стеатореяДефицит жирорастворимых витаминовОтеки (гипопротеинемия)Задержка физического развитияВыпадение")
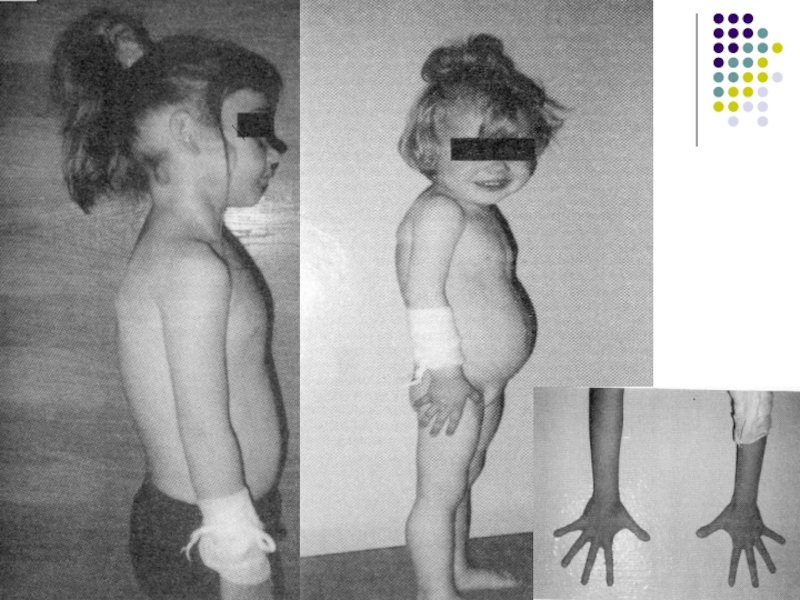
Слайд 37Осложнение - бесплодие
Случаи полного бесплодия у женщин (например, полное отсутствие овуляции)
крайне редки, чаще проблема в вязкой слизи, которая затрудняет движение спермы.
95% больных МВ мужчин страдают бесплодием. Основная причина – полная закупорка семявыводящих канальцев.
95% больных МВ мужчин страдают бесплодием. Основная причина – полная закупорка семявыводящих канальцев.
 крайне редки, чаще проблема")
Слайд 39Генетический анализ (медико-генетическое консультирование)
Пренатальная диагностика
Неонатальная диагностика
Постнатальная диагностика – характерные клинические
симптомы, потовый тест, ФВД, копрограмма, посев мокроты, рентгенограмма ОГК, УЗИ ОБП, ЭКГ, сатурация кислородом, антропометрия 1 раз в 3 мес
Тест на фертильность
Тест на фертильность
Возможности диагностики
Пренатальная диагностика Неонатальная диагностикаПостнатальная диагностика – характерные клинические симптомы, потовый тест, ФВД,")
Слайд 40Потовый тест
Нужно не менее 100мг потовой жидкости, повторяется минимум трижды
Начинать исследование
целесообразно при достижении массы не менее 3кг
Диагностический критерий – выше 60ммоль/л у детей и 70ммоль/л у подростков и взрослых
У здоровых лиц концентрация натрия обычно выше, чем хлора, у больных М – хлора больше, чем натрия
Сумма ионов натрия и хлора у здоровых меньше 140, а у больных М больше 140 ммоль/л
Диагностический критерий – выше 60ммоль/л у детей и 70ммоль/л у подростков и взрослых
У здоровых лиц концентрация натрия обычно выше, чем хлора, у больных М – хлора больше, чем натрия
Сумма ионов натрия и хлора у здоровых меньше 140, а у больных М больше 140 ммоль/л

Слайд 41Диета
Основной принцип – повышение энергетической ценности пищи на 20-25%:
жиры -
40-45%, белки - 15% и углеводы - 45-50%;
-увеличение полиненасыщенных жиров;
-дополнительное подсаливание пищи
Оптимальное вскармливание – грудное, искусственные смеси, содержащие среднецепочечные жирные кислоты
-увеличение полиненасыщенных жиров;
-дополнительное подсаливание пищи
Оптимальное вскармливание – грудное, искусственные смеси, содержащие среднецепочечные жирные кислоты
Лечение

Слайд 42Заместительная терапия - ферменты
Должна быть пожизненной, беспрерывной, достаточной, с физиологическим соотношением
липазы, протеазы, амилазы
Дозирование: детям раннего возраста около 4000 МЕ липазы на 100-150мл молока; старше 1 года – 2000-6000 МЕ/кг в сутки
Дозирование: детям раннего возраста около 4000 МЕ липазы на 100-150мл молока; старше 1 года – 2000-6000 МЕ/кг в сутки

Слайд 43Антибактериальная терапия
До 2 лет жизни ребенка наиболее частой бактериальной флорой являются
St.aureus, Haemophilus influenzae. С возрастом частота выделения Ps.aeruginosa увеличивается.
Профилактическое в/в назначение антибиотикотерапии регулярными курсами независимо от клиники с определенными интервалами (1 раз в 3-4 месяца) в/в
Наиболее широко используются:
Азлоциллин, Цефтазидим, Азтреонам , Тиенам, Гентамицин, тобрамицин
Профилактическое в/в назначение антибиотикотерапии регулярными курсами независимо от клиники с определенными интервалами (1 раз в 3-4 месяца) в/в
Наиболее широко используются:
Азлоциллин, Цефтазидим, Азтреонам , Тиенам, Гентамицин, тобрамицин

Слайд 44Ацетилцистеин (ингаляци, сироп)
Амбробене (ингаляци, сироп)
Рекомбинантная ДНК-аза «Пульмозим»
Физические методы эвакуации мокроты
Постуральный дренаж
Аутогенный
дренаж – с помощью дыхательных движений во время вдоха и выдоха
ЛФК с активным циклом дыхания «хаффинг» - чередование спокойных циклов с форсированным выдохом в течение 10-15мин
ЛФК с активным циклом дыхания «хаффинг» - чередование спокойных циклов с форсированным выдохом в течение 10-15мин
Муколитики
Амбробене (ингаляци, сироп)Рекомбинантная ДНК-аза «Пульмозим»Физические методы эвакуации мокротыПостуральный дренажАутогенный дренаж – с помощью")






