- Главная
- Разное
- Дизайн
- Бизнес и предпринимательство
- Аналитика
- Образование
- Развлечения
- Красота и здоровье
- Финансы
- Государство
- Путешествия
- Спорт
- Недвижимость
- Армия
- Графика
- Культурология
- Еда и кулинария
- Лингвистика
- Английский язык
- Астрономия
- Алгебра
- Биология
- География
- Детские презентации
- Информатика
- История
- Литература
- Маркетинг
- Математика
- Медицина
- Менеджмент
- Музыка
- МХК
- Немецкий язык
- ОБЖ
- Обществознание
- Окружающий мир
- Педагогика
- Русский язык
- Технология
- Физика
- Философия
- Химия
- Шаблоны, картинки для презентаций
- Экология
- Экономика
- Юриспруденция
Генные и хромосомные болезни презентация
Содержание
- 1. Генные и хромосомные болезни
- 2. Генные болезни- это разнообразная по клинической картине
- 3. По типу наследования генные заболевания распределяют на
- 4. Аутосомно - доминантные заболевания. Синдром Марфана .
- 5. Синдром Марфана. Аутосомно-доминантное заболевание из группы наследственных
- 6. Без лечения продолжительность жизни лиц с синдромом
- 7. Нейрофиброматоз. Самое распространённое наследственное заболевание, предрасполагающее к
- 8. В половине случаев заболевание является наследственным, в
- 9. Ахондроплазия – аутосомно-доминантное заболевание.
- 10. Синдром Холт- Орама. наследственное сочетание аномалий больших
- 11. Чаще поражается левая рука. Наблюдаются и другие
- 12. прогерия Прогерия (греч. преждевременно состарившийся) — патологическое
- 13. Прогерия (синдром Гетчинсона-Гилфорда) аутосомно-доминантное заболевание. Клинические
- 14. Аутосомно-рецессивные заболевания. Муковисцидоз(Кистофиброз поджелудочной железы) Фенилкетонурия(Фенилпировиноградная олигофрения) Адреногенитальный синдром(Врожденная гиперплазия коры надпочечников)
- 15. Муковисцидоз. Системное наследственное заболевание, обусловленное мутацией гена
- 16. Патологические изменения в лёгких характеризуются признаками хронического
- 18. Муковисцидоз - аутосомно-рецессивное
- 19. Фенилкетонурия. наследственное заболевание группы ферментопатий, связанное с
- 20. Диагностика производится полуколичественным тестом или количественным определением
- 21. Фенилкетонурия – аутосомно-рецессивное заболевание. Причина
- 22. Галактоземия - аутосомно-рецессивное заболевание.
- 23. Альбинизм – аутосомно-рецессивное заболевание. Причина
- 24. Андреногенитальный синдром. группа заболеваний, наследуемых по аутосомно-рецессивному
- 25. Х- сцепленные рецессивные заболевания. Псевдогипертрофическая мышечная дистрофия
- 26. Псевдогипертрофическая мышечная дистрофия Дюшенна. Мышечная дистрофия —врожденное
- 27. Мышечная дистрофия развивается из-за постепенного разрушения нервно-мышечных
- 28. Синдром Мартина- Белл. Синдром с ломкой Х
- 29. Фенотипические признаки: большая голова с высоким и
- 30. Гемофилия- нарушение свёртываемости крови.
- 31. Гемофилия - Х-сцепленное рецессивное заболевание. Частота:
- 32. Гемофилия. Гемартрозы коленных суставов (а) и стоп (б)
- 33. Гидроцефалия - Х-сцепленное рецессивное заболевание.
- 34. Дальтонизм является одной из наиболее распространенных
- 35. XN XN XN Xn XN y Xn y Дальтонизм N = норма n = дальнонизм
- 36. Y-сцепленный тип: 1) болеют только мужчины; 2)
- 37. Гипертрихоз ушных раковин - Y-сцепленный признак
- 38. Сравнение аутосомных и сцепленных с полом генных заболеваний
- 39. Митохондриальные болезни Каждая митохондрия
- 40. Хромосомные болезни. К хромосомным относятся болезни, обусловленные
- 41. Аутосомные трисомии. Синдром Дауна(Болезнь Дауна) Синдром Патау Синдром Эдвардса
- 42. Синдром Дауна. одна из форм геномной патологии,
- 43. Возраст матери влияет на шансы зачатия ребёнка
- 44. «плоское лицо» — 90 % брахицефалия (аномальное
- 45. Синдром Патау. Хромосомное заболевание человека, которое характеризуется
- 46. При синдроме Патау наблюдаются тяжелые врожденные пороки.
- 47. У 80 % новорожденных встречаются пороки развития
- 48. Синдром Эдвардса. хромосомное заболевание, характеризуется комплексом множественных
- 49. Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего
- 50. Из дефектов внутренних органов наиболее часто отмечаются
- 51. Полисомии по половым хромосомам. Синдром трисомии Х
- 52. Синдром Трисомии Х. Заболевание встречается сравнительно редко
- 53. Для заболевания характерно лишь небольшое снижение интеллекта.
- 54. Синдром Клайнфельтера. Генетической особенностью этого синдрома является
- 55. До начала полового развития удается отметить только
- 56. В подростковом возрасте синдром чаще всего проявляется
- 57. Синдром дисомии по Y-хромосоме. Частотой 1 на
- 58. Вообще в заболевании нет ничего особенного, Y-хромосома
- 59. Синдром Шерешевского-Тёрнера. Хромосомная болезнь, сопровождающаяся характерными
- 60. Половое недоразвитие при синдроме Тернера отличается определённым
- 61. Интеллект у большинства больных с синдромом Тернера
- 62. Синдромы частичных моносомий. Синдром «Кошачьего крика»
- 63. Хромосомно синдром кошачьего крика объясняется частичной моносомией;
- 64. При этом синдроме наблюдается: общее отставание в
- 65. Спасибо за внимание!

Слайд 2Генные болезни- это разнообразная по клинической картине группа заболеваний , обусловленная
В результате мутации гена на молекулярном уровне возможны следующие варианты:
синтез аномального белка;
выработка избыточного количества генного продукта;
отсутствие выработки первичного продукта;
выработка уменьшенного количества нормального первичного продукта.

Слайд 3По типу наследования генные заболевания распределяют на группы:
Аутосомно-доминантные
Аутосомно-рецессивные
Х-сцепленные доминантные
Х-сцепленные рецессивные
Y-сцепленные
Для диагностики генных заболеваний используют биохимический, генеалогический методы генетики и метод амниоцентеза.

Слайд 4Аутосомно - доминантные заболевания.
Синдром Марфана .
Нейрофиброматоз (Болезнь Реклингхаузена ).
Синдром Холт -
.Синдром Холт - Орама (Рука-сердце)")
Слайд 5Синдром Марфана.
Аутосомно-доминантное заболевание из группы наследственных патологий соединительной ткани. Синдром вызван

Слайд 6Без лечения продолжительность жизни лиц с синдромом Марфана часто ограничивается 30—40

Слайд 7Нейрофиброматоз.
Самое распространённое наследственное заболевание, предрасполагающее к возникновению опухолей у человека. Описан

Слайд 8В половине случаев заболевание является наследственным, в половине — результатом спонтанной
Для заболевания характерно появление множественных пигментированных пятен цвета «кофе с молоком», доброкачественных новообразований — нейрофибром, опухолей центральной нервной системы, костных аномалий, изменений радужной оболочки глаза и целого ряда других симптомов.

Слайд 9 Ахондроплазия – аутосомно-доминантное заболевание.
Частота: 1 :
Причина – нарушение роста трубчастых костей. Основные проявления: карликовость с короткими конечностями, большой головой, нормальным туловищем, лордозом. Интелект, як правило, нормальный. Репродуктивная способность не нарушена.

Слайд 10Синдром Холт- Орама.
наследственное сочетание аномалий больших пальцев рук и дефекта межпредсердной

Слайд 11Чаще поражается левая рука. Наблюдаются и другие скелетные изменения: гипоплазия лопаток

Слайд 12прогерия
Прогерия (греч. преждевременно состарившийся) — патологическое состояние, характеризующееся комплексом изменений кожи,
 — патологическое состояние, характеризующееся комплексом изменений кожи, внутренних органов, обусловленных преждевременным")
Слайд 13Прогерия (синдром Гетчинсона-Гилфорда)
аутосомно-доминантное заболевание. Клинические проявления: прогрессирующее, быстрое старение организма
аутосомно-доминантное заболевание. Клинические проявления: прогрессирующее, быстрое старение организма с 5-6-летнего возраста. Больные умирают")
Слайд 14Аутосомно-рецессивные заболевания.
Муковисцидоз(Кистофиброз поджелудочной железы)
Фенилкетонурия(Фенилпировиноградная олигофрения)
Адреногенитальный синдром(Врожденная гиперплазия коры надпочечников)
Фенилкетонурия(Фенилпировиноградная олигофрения)Адреногенитальный синдром(Врожденная гиперплазия коры надпочечников)")
Слайд 15Муковисцидоз.
Системное наследственное заболевание, обусловленное мутацией гена трансмембранного регулятора муковисцидоза и характеризующееся

Слайд 16Патологические изменения в лёгких характеризуются признаками хронического бронхита с развитием бронхоэктазов
В поджелудочной железе выявляется диффузный фиброз, утолщение междольковых соединительнотканных прослоек, кистозные изменения мелких и средних протоков. В печени отмечается очаговая или диффузная жировая и белковая дистрофия клеток печени, желчные стазы в междольковых желчных протоках, лимфогистиоцитарные инфильтраты в междольковых прослойках, фиброзная трансформация и развитие цирроза.
При мекониевой непроходимости выражена атрофия слизистого слоя, просвет слизистых желез кишечника расширен, заполнен эозинофильными массами секрета, местами имеет место отёк подслизистого слоя, расширение лимфатических щелей. Нередко муковисцидоз сочетается с различными пороками развития желудочно-кишечного тракта.

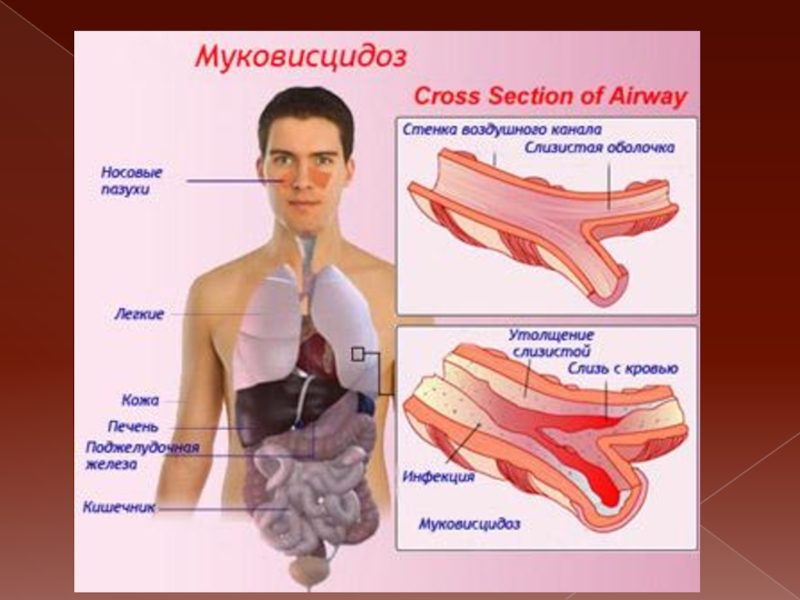
Слайд 18
Муковисцидоз - аутосомно-рецессивное заболевание
Частота: 1 : 2 500 новорожденых.
Клинические формы: 1) смешанная (поражение дыхательной и пищеварительной систем;
2) легочная; 3) кишечная; 4) печеночная; 5) электролитная (поражение поджелудочной железы).
Диагностика 1) потовая проба (увеличение натрия хлорида в поте); 2) наличие трипсина в кале; 3) ДНК-диагностика.
. Лечение включает ферменты поджелудочной железы, муколитики

Слайд 19Фенилкетонурия.
наследственное заболевание группы ферментопатий, связанное с нарушением метаболизма аминокислот, главным образом

Слайд 20Диагностика производится полуколичественным тестом или количественным определением фенилаланина в крови. При

Слайд 21 Фенилкетонурия – аутосомно-рецессивное заболевание. Причина – недостаток фермента фенилаланин-4-моноксидазы.

Слайд 22 Галактоземия - аутосомно-рецессивное заболевание.
Причина – недостаток фермента
Симптомы заболевания проявляются у новорожденных после приема молока. Характеризуется увеличенной печенью, рвотой, поносами, умственной отсталостью. Лечение состоит в выключении молока из пищи.

Слайд 23 Альбинизм – аутосомно-рецессивное заболевание. Причина – отсутствие фермента тирозиназы,

Слайд 24Андреногенитальный синдром.
группа заболеваний, наследуемых по аутосомно-рецессивному пути, при которых нарушается выработка
У девочек возникает незначительная гиперплазия клитора , ускорение костного возраста, различные нарушения менструального цикла.
У мальчиков могут быть преждевременное оволосение наружных гениталий и снижение темпов роста за счет раннего закрытия зон роста.

Слайд 25Х- сцепленные рецессивные заболевания.
Псевдогипертрофическая мышечная дистрофия Дюшенна.
Синдром умственной отсталости с ломкой
")
Слайд 26Псевдогипертрофическая мышечная дистрофия Дюшенна.
Мышечная дистрофия —врожденное заболевание, характеризующееся мышечной слабостью.
Синдром Дюшенна

Слайд 27Мышечная дистрофия развивается из-за постепенного разрушения нервно-мышечных связей.
Это врожденное

Слайд 28Синдром Мартина- Белл.
Синдром с ломкой Х хромосомы - сцепленное с полом
Главным симптомом синдрома является интеллектуальное недоразвитие и своеобразная речь. Такие больные говорят быстро, сбивчиво, имеются персеверации (бормочущая речь). Также могут быть нарушения поведения в виде агрессивности, двигательной расторможенности. В качестве одной из частых психопатологических особенностей отмечена шизофреноподобная симптоматика, включающая в себя подпрыгивания, похлопывания руками, повороты вокруг своей оси, встряхивание кистями, «манежный» бег, разнообразные гримасы, монотонное хныканье.

Слайд 29Фенотипические признаки: большая голова с высоким и широким лбом, длинное лицо
Неврологическая симптоматика неспецифична, определяется как и у всех детей с умственной отсталостью. Наблюдается некоторая мышечная гипотония, дискоординация движений.

Слайд 30
Гемофилия- нарушение свёртываемости крови.
Первоначальной носительницей мутантного гена

Слайд 31Гемофилия - Х-сцепленное рецессивное заболевание.
Частота: 1:2500 новорожденных. Характеризуется кровотечениями, гемартрозами
. Причина")
 и стоп (б)")
Слайд 33 Гидроцефалия - Х-сцепленное рецессивное заболевание. Частота: 1 : 2000
Причина – нарушение оттока спинномозковой жидкости. Характеризуется увеличением размеров головы, неврологическими расстройствами, умственной отсталостью.

Слайд 34Дальтонизм
является одной из наиболее распространенных аномалий, которые наследуются рецессивно, сцепленно
Характеризуется нарушением восприятия красного и зеленого цветов.


 болеют только мужчины;2) отец передает признак всем сыновьям.")


Слайд 39Митохондриальные болезни
Каждая митохондрия имеет собственную ДНК кольцевой формы. В
Генные мутации в митохондриальной ДНК наблюдаются при наследственной атрофии зрительного нерва Лебера, митохондриальных миопатиях, при миокардиопатиях, атаксии-слепоте. Митохондрии передаются с цитоплазмой яйцеклеток, сперматозоиды цитоплазмы почти не содержат.
Для митохондрального наследования характерны следующие признаки:
1) болезнь передается только от матери детям;
2) болеют и девочки, и мальчики;
3) больной отец не передает заболевания ни дочерям, ни сыновьям.
 содержится 16569")
Слайд 40Хромосомные болезни.
К хромосомным относятся болезни, обусловленные геномными мутациями или структурными изменениями
Все хромосомные болезни принято делить на две группы: аномалии числа хромосом и нарушения структуры хромосом.

Синдром ПатауСиндром Эдвардса")
Слайд 42Синдром Дауна.
одна из форм геномной патологии, при которой чаще всего кариотип

Слайд 43Возраст матери влияет на шансы зачатия ребёнка с синдромом Дауна. Если

Слайд 44«плоское лицо» — 90 %
брахицефалия (аномальное укорочение черепа) — 81 %
кожная
Эпикантус- 80%
гиперподвижность суставов — 80 %
мышечная гипотония — 80 %
плоский затылок — 78 %
короткие конечности — 70 %
брахимезофалангия (укорочение всех пальцев за счёт недоразвития средних фаланг) — 70 %
катаракта в возрасте старше 8 лет — 66 %
открытый рот (в связи с низким тонусом мышц и особым строением нёба) — 65 %
зубные аномалии — 65 %
клинодактилия 5-го пальца (искривлённый мизинец) — 60 %
плоская переносица — 52 %
бороздчатый язык — 50 %
поперечная ладонная складка (называемая также «обезьяньей») — 45 %
короткая широкая шея — 45 %
ВПС (врождённый порок сердца) — 40 %
короткий нос — 40 %
страбизм (косоглазие) — 29 %
деформация грудной клетки, килевидная или воронкообразная — 27 %
стеноз или атрезия двенадцатиперстной кишки — 8 %
врождённый лейкоз — 8 %.
 — 81 %кожная складка на шее у")
Слайд 45Синдром Патау.
Хромосомное заболевание человека, которое характеризуется наличием в клетках дополнительной хромосомы
Характерным осложнением беременности при вынашивании плода с синдромом Патау является многоводие: оно встречается почти в 50 % случаев Синдрома Патау.
Встречается с частотой 1:7000-1:14000. Имеются два цитогенетических варианта синдрома Патау: простая трисомия и робертсоновская транслокация.

Слайд 46При синдроме Патау наблюдаются тяжелые врожденные пороки. Дети с синдромом Патау

Слайд 47У 80 % новорожденных встречаются пороки развития сердца: дефекты межжелудочковой и
В связи с тяжелыми врожденными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы (95 % — до 1 года).

Слайд 48Синдром Эдвардса.
хромосомное заболевание, характеризуется комплексом множественных пороков развития и трисомией 18
Популяционная частота примерно 1:7000. Дети с трисомией 18 чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21 и 13. Для женщин старше 45 лет риск родить больного ребёнка составляет 0,7 %. Девочки с синдромом Эдвардса рождаются в три раза чаще мальчиков.

Слайд 49Фенотипические проявления синдрома Эдвардса многообразны. Чаще всего возникают аномалии мозгового и

Слайд 50Из дефектов внутренних органов наиболее часто отмечаются пороки сердца и крупных
Продолжительность жизни детей с синдромом Эдвардса невелика: 60% детей умирают в возрасте до 3 мес, до года доживает лишь 5-10 %. Основной причиной смерти служат остановка дыхания и нарушения работы сердца. Оставшиеся в живых — глубокие олигофрены.

Слайд 51Полисомии по половым хромосомам.
Синдром трисомии Х
Синдром Клайнфельтера
Синдром дисомии по Y- хромосоме
Синдром

Слайд 52Синдром Трисомии Х.
Заболевание встречается сравнительно редко (1 случай на 700—800 новорожденных
Часто трисомия-Х выявляется у больных психоза В ряде случаев могут развиться вторичная аменорея, преждевременный климакс. У большинства больных менструальный цикл не нарушен. При нормальной функции яичников женщины с трисомией-Х способны к деторождению. По наследству трисомия-Х не передается. ми и олигофренией.
Прогноз для жизни благоприятный. В ряде случаев у таких женщин сохранена детородная функция.
. Часто трисомия-Х выявляется")
Слайд 53Для заболевания характерно лишь небольшое снижение интеллекта. В ряде случаев интеллект
Иногда наблюдаются патологические изменения органа зрения: двусторонняя атрофия зрительного нерва, хориоретинит, одностороннее помутнение роговицы.

Слайд 54Синдром Клайнфельтера.
Генетической особенностью этого синдрома является разнообразие цитогенетических вариантов и их
. Обнаружено несколько")
Слайд 55До начала полового развития удается отметить только отдельные физические признаки, такие
К началу полового созревания формируются характерные пропорции тела: больные часто оказываются выше сверстников, но в отличие от типичного евнухоидизма размах рук у них редко превышает длину тела, ноги заметно длиннее туловища. Кроме того, некоторые дети с данным синдромом могут испытывать трудности в учёбе и в выражении своих мыслей. В некоторых руководствах указывается, что у пациентов с синдромом Клайнфельтера отмечается несколько сниженный объём яичек до периода полового созревания. Это утверждение является неверным, поскольку до периода полового созревания объём яичек у всех мальчиков небольшой — менее 1 мл

Слайд 56В подростковом возрасте синдром чаще всего проявляется увеличением грудных желез, хотя

Слайд 57Синдром дисомии по Y-хромосоме.
Частотой 1 на 1000 новорожденных мальчиков.
Мужчины с
Иногда наблюдается незначительное снижение интеллекта, склонность к агрессивным и антисоциальным поступкам. По некоторым данным, в местах заключения мужчин с генотипом XYY в 10 раз больше, чем мужчин с нормальным генотипом.

Слайд 58Вообще в заболевании нет ничего особенного, Y-хромосома — содержит ген SRY,
У большинства больных отмечается ускорение роста в детском возрасте .
В 30 — 40 % случаев отмечаются определенные симптомы — грубые черты лица, выступающие надбровные дуги и переносица, увеличенная нижняя челюсть, высокое нёбо, аномальный рост зубов с дефектами зубной эмали, большие ушные раковины, деформация коленных и локтевых суставов. Интеллект или негрубо снижен, или в норме. Характерны эмоционально-волевые нарушения: агрессивность, взрывчатость, импульсивность. В то же время для этого синдрома характерны подражательность, повышенная внушаемость, причем больные наиболее легко усваивают негативные формы поведения.

Слайд 59Синдром Шерешевского-Тёрнера.
Хромосомная болезнь, сопровождающаяся характерными аномалиями физического развития, низкорослостью и
При синдроме Тернера патологические признаки по частоте встречаемости распределяются следующим образом: низкорослость (98%), общая диспластичность (неправильное телосложение) (92%), бочкообразная грудная клетка (75%), укорочение шеи (63%), низкий рост волос на шее (57%), высокое «готическое» нёбо (56%), крыловидные складки кожи в области шеи (46%), деформация ушных раковин (46%), укорочение метакарпальных и метатарзальных костей и аплазия фаланг (46%), деформация локтевых суставов (36%), множественные пигментные родинки (35%), лимфостаз (24%), пороки сердца и крупных сосудов (22%), повышенное артериальное давление (17%).

Слайд 60Половое недоразвитие при синдроме Тернера отличается определённым своеобразием. Нередкими признаками являются

Слайд 61Интеллект у большинства больных с синдромом Тернера практически сохранен, однако частота
Прогноз для жизни при синдроме Тернера благоприятный, исключение составляют больные с тяжёлыми врождёнными пороками сердца и крупных сосудов и почечной гипертензией. Лечение женскими половыми гормонами делает больных способными к семейной жизни, однако абсолютное большинство из них остаются бесплодными.


Слайд 63Хромосомно синдром кошачьего крика объясняется частичной моносомией; он развивается при делеции

Слайд 64При этом синдроме наблюдается:
общее отставание в развитии,
низкая масса при рождении и
лунообразное лицо с широко расставленными глазами
характерный плач ребёнка, напоминающий кошачье мяуканье, причиной которого является изменение гортани (сужение, мягкость хрящей, уменьшение надгортанника, необычная складчатость слизистой оболочки) или недоразвитие гортани. Признак исчезает к концу первого года жизни.
Частота синдрома примерно 1:45000.
Клиническая картина синдрома и продолжительность жизни людей с этим синдромом довольно сильно варьирует по сочетанию врождённых пороков развития органов.







