- Главная
- Разное
- Дизайн
- Бизнес и предпринимательство
- Аналитика
- Образование
- Развлечения
- Красота и здоровье
- Финансы
- Государство
- Путешествия
- Спорт
- Недвижимость
- Армия
- Графика
- Культурология
- Еда и кулинария
- Лингвистика
- Английский язык
- Астрономия
- Алгебра
- Биология
- География
- Детские презентации
- Информатика
- История
- Литература
- Маркетинг
- Математика
- Медицина
- Менеджмент
- Музыка
- МХК
- Немецкий язык
- ОБЖ
- Обществознание
- Окружающий мир
- Педагогика
- Русский язык
- Технология
- Физика
- Философия
- Химия
- Шаблоны, картинки для презентаций
- Экология
- Экономика
- Юриспруденция
Технология получения полимеров презентация
Содержание
- 1. Технология получения полимеров
- 2. Введение (цели и задачи курса) Данный курс
- 3. Практически всё, что окружает нас: одежда, обувь,
- 4. Основные этапы формирования Химии высокомолекулярных соединений как самостоятельной науки
- 6. ПОЛИМЕРНОЕ СОСТОЯНИЕ ВЕЩЕСТВА Полимер – это вещество, состоящее из
- 7. Классификация полимеров по происхождению природные: натуральный каучук,
- 8. Классификация полимеров по областям применения пластические
- 9. Ассортимент мирового производства крупнотоннажных полимеров -
- 10. Мир Европа Динамика производства пластиков млн. т
- 11. МИРОВОЕ ПОТРЕБЛЕНИЕ ПЛАСТИКОВ в 2007 – 2015 г.г.
- 12. Номенклатура полимеров Составные части макромолекул Составные звенья
- 13. Наименьшее составное звено, повторением которого может быть
- 14. Основные виды номенклатуры Номенклатура,
- 15. Номенклатура полимеров
- 17. 2 Введение в теорию полимеризационных процессов
- 18. Основные параметры процесса ПМ - термодинамические
- 20. Для производных этилена 2⋅ЕС-С > ЕС=С, поэтому
- 21. где ΔGо = - RTlnKp - стандартный
- 22. Кинетические параметры Кинетические параметры позволяют описать
- 23. Длина цепи Полимеризация вызывается первичными активными центрами,
- 24. Элементарные стадии процесса полимеризации Полимеризационные процессы в
- 25. Методы проведения процесса Полимеризация мономера может
- 26. Радикальная полимеризация Радикальная полимеризация – полимеризация, инициируемая
- 28. Мономеры, способные вступать в реакцию радикальной полимеризации
- 29. Введение заместителей в молекулу бутадиена приводит к
- 30. Относительные скорости полимеризации различных диеновых мономеров
- 31. Мономеры с неявной системой сопряжения двойных связей
- 32. Механизм радикальной полимеризации Радикальная полимеризация инициируется свободными
- 33. Инициирование Инициирование - образование активных центров в
- 34. Гомолитический распад активируется под действием тепла, света,
- 35. Для перевода молекул в активное состояние существуют
- 36. Инициирование под действием физических факторов Термическое инициирование Мономолекулярный механизм инициирования Штаудингера Бимолекулярный механизм инициирования
- 37. Экспериментально установлено, что растущая полимерная цепь -
- 38. Фотохимическое инициирование Инициирование связано с поглощением молекулой
- 39. Радиационно-химическое инициирование Протекает под воздействием ионизирующих излучений
- 40. Химическое инициирование Химическое инициирование является наиболее распространенным
- 41. В качестве инициаторов используются следующие классы соединений:
- 44. Период полураспада (τ1/2): Распад пероксида
- 45. Распад динитрила азо(бис)изомасляной кислоты
- 46. Эффект клетки (эффект Франко-Рабиновича) Уменьшение количества свободных
- 47. 3. Выход из клетки: - за
- 48. Окислительно-восстановительное инициирование 1 При введении в реакционную
- 49. Системы, приводящие к образованию одиночного радикала -
- 51. В данном рецепте Fe2+ может использоваться в
- 52. Первичные свободные радикалы, образовавшиеся по любому из
- 53. В ходе реакции происходит выделение тепла.
- 54. Стадия обрыва полимерной цепи
- 55. 3. Столкновении свободных полимерных
- 56. При этом могут возникнуть
- 57. Способность к участию в
- 58. Передача цепи на мономер
- 59. Бис(изопропилксантоген)дисульфид, дипроксид CBr4
- 60. Передача цепи на инициатор
- 61. Передача цепи на полимер
- 63. Реакции замедления и ингибирования
- 64. Время задержки процесса полимеризации
- 65. Механизм действия замедлителей и
- 66. Образующийся хинон взаимодействует со
- 67. Замедлители по своему действию
- 68. Роль кислорода в процессах
- 69. Реакции разветвления и перекрещивания
- 70. Некоторые особенности полимеризации диенов
- 71. Аутополимеризация (самопроизвольная
- 72. Передача цепи на
- 73. Гель-эффект (эффект Тромсдорфа) Время
- 74. Кинетический анализ реакции
- 75. Концентрацию радикалов,
- 76. Длина кинетической цепи
- 77. Во многих случаях
- 78. С учетом реакций
- 79. Например, отношение
- 80. Влияние различных факторов
- 81. Энергии активации и константы
- 82. Влияние инициатора С увеличением
- 83. Способы проведения радикальной полимеризации
- 84. Полимеризация в растворе Отсутствуют
- 85. Полимеризация в суспензии (Бисерная
- 86. Твердофазная полимеризация
- 87. Полимеризация в эмульсии
- 88. Эмульсионная радикальная полимеризация
- 89. сополимеры хлоропрена и
- 90. Достоинства эмульсионной полимеризации:
- 91. Недостатки: Невозможность
- 92. Механизм эмульсионной полимеризации
- 93. Трудность создания единой
- 94. Минимальная концентрация эмульгатора,
- 95. С17Н35 СООК
- 96. С мицеллообразованием тесно
- 97. В том случае,
- 98. По ходу полимеризации
- 99. Кинетическая кривая процесса
- 100. На мицеллярном этапе:
- 101. Стационарный этап:
- 102. Теория Медведева-Хомяковского Согласно
- 103. СОПОЛИМЕРИЗАЦИЯ Сополимеризацией называют
- 104. Допущения: - постоянная
- 105. Скорости исчерпания мономеров
- 106. В стационарном состоянии
- 107. Параметры r1 и
- 108. Таким образом химический
- 110. Статистический анализ чередования
- 111. Дифференциальный и интегральный составы
- 112. Изменение дифференциального и
- 113. Константы сополимеризации и
- 114. ИОННАЯ ПОЛИМЕРИЗАЦИЯ Ионная
- 115. Каждая из форм
- 116. Внедрение мономера в растущую полимерную цепь осуществляется
- 117. Существенный характер имеет
- 118. КАТИОННАЯ ПОЛИМЕРИЗАЦИЯ Впервые
- 119. Катализаторы: - протонные
- 120. Реакция инициирования заключается
- 121. Рост цепи в
- 122. Обрыв цепи чаще
- 123. Энергия активации этого
- 124. Иногда в процессе
- 125. Отсюда следует, что
- 126. АНИОННАЯ ПОЛИМЕРИЗАЦИЯ Систематическое
- 127. Реакция инициирования может
- 128. или между металлом
- 129. Особенностью анионной полимеризации
- 130. Полимеризация неполярных мономеров
- 131. Рост цепи для
- 132. В наибольшей степени
- 133. Чаще всего общей
- 134. Общая кинетическая картина
- 135. Часто наблюдаются более
- 136. ИОННО-КООРДИНАЦИОННАЯ ПОЛИМЕРИЗАЦИЯ Если
- 137. Добавление всего лишь
- 138. В этом случае
- 139. В 1955 году
- 140. Типичными катализаторами ионо-координационнной
- 141. В акте координации
- 142. Стереоспецифичность каталитических систем
- 143. Скорость роста цепи
- 144. ПОЛИКОНДЕНСАЦИЯ Поликонденсация -
- 145. Большой вклад в
- 146. КЛАССИФИКАЦИЯ И ТЕРМИНОЛОГИИ
- 147. По типу (и
- 148. При поликонденсации часто
- 149. 1) Мономеры (например,
- 150. В условиях поликонденсации
- 151. Примаер: при поликонденсации тетрафункциональных мономеров -бис-(о-фенилендиаминов) с
- 152. Часто ограничиваются рассмотрением
- 153. Например, при получении
- 154. При этом взаимодействие
- 155. Таким образом росту
- 156. Возможность циклизации определяется
- 157. В результате совместного
- 158. В ряде случаев
- 159. КИНЕТИКА, КАТАЛИЗ, МОЛЕКУЛЯРНО-МАССОВОЕ
- 160. Поэтому особенно важно
- 161. Во многих случаях
- 162. Кинетическая и другие
- 163. Из этого соотношения
- 164. Процессы поликонденсации можно
- 165. - введение активирующих
- 166. Катализаторы применяют как
- 167. В этом случае
- 168. Таким образом
- 169. При х =

Слайд 2Введение (цели и задачи курса)
Данный курс содержит основные теоретические представления о
В частности, в нем рассматриваются механизмы синтеза основных полимеров, выпускаемых в мире, а также вопросы технологии их получения и их свойства.
Данный курс содержит основные теоретические представления о различных типах реакций полимеризации.В")
Слайд 3Практически всё, что окружает нас: одежда, обувь, мебель, продукты питания, книги,
Кроме того, возникновение жизни на Земле обусловлено появлением простейших органических полимерных соединений в "первичном бульоне".


Слайд 6ПОЛИМЕРНОЕ СОСТОЯНИЕ ВЕЩЕСТВА
Полимер – это вещество, состоящее из множества молекул большой молекулярной массы
1. Основные понятия
.")
Слайд 7Классификация полимеров по происхождению
природные: натуральный каучук, целлюлоза, крахмал, хитин, белки и
синтетические полимеры (не существующие в природе): полиэтилен, полипропилен, полистирол, полибутадиен и др.
- искусственные, получаемые модификацией природных полимеров: нитроцеллюлоза, метилцеллюлоза, ацетатцеллюлоза и др.

Слайд 8Классификация полимеров по областям применения
пластические массы - полимеры, у которых
каучуки (Тст<Ткомн): полибутадиен, полиизобутилен и др.
синтетические и искусственные волокна: полиакрилонитрил, полиэтилентерефталат, поликапролактам, найлон-66, вискоза и др.
плёнки: лаки, краски, эмали
- биологические полимеры (биополимеры)

Слайд 9
Ассортимент мирового производства крупнотоннажных полимеров
- крупнотоннажные,
- среднетоннажные,
- мелкотоннажные
Классификация полимеров



Слайд 12Номенклатура полимеров
Составные части макромолекул
Составные звенья (СЗ) – это все возможные фрагменты, т.е.
 – это все возможные фрагменты, т.е. атомы или группы атомов,")
Слайд 13Наименьшее составное звено, повторением которого может быть описано строение регулярного полимера,
Составные части макромолекул

Слайд 14Основные виды номенклатуры
Номенклатура, основанная на названии мономеров (рациональная
Название формируется путем прибавления приставки "поли" (в случае нерганического полимера - “катена-поли-”) к названию мономера.
Номенклатура, основанная на химической структуре полимерной цепи (систематическая номенклатура IUPAC).
Название формируется путем прибавления приставки "поли" к названию составного повторяющегося звена по номенклатуре IUPAC с учетом расположения и ориентации этого звена в цепи.
Случайная номенклатура не подразумевает каких-либо закономерностей в образовании названий полимеров, но ИЮПАК рекомендует использовать ее для обозначения наиболее часто применяемых полимеров.
. Название формируется путем")


Слайд 172 Введение в теорию полимеризационных процессов
Основные типы процесса синтеза полимеров
поликонденсация (иногда называемая ступенчатой полимеризацией) - макромолекулы соединяются путем реакций функциональных групп
Полимеризацией называется реакция, в ходе которой молекулы мономера последовательно присоединяются к активному центру, находящемуся на конце растущей цепи
Основные типы:
- радикальная полимеризация,
ионная полимеризация,
ионно-координационная полимеризация.

Слайд 18Основные параметры процесса ПМ
- термодинамические
- кинетические
Термодинамические параметры
Термодинамические параметры характеризуют возможность
Термодинамическое условие протекания реакции полимеризации

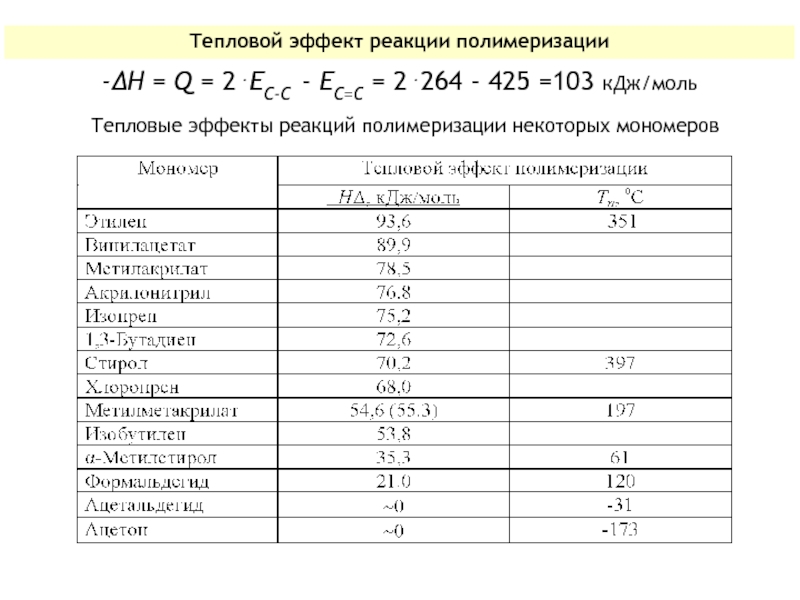
Слайд 20Для производных этилена 2⋅ЕС-С > ЕС=С, поэтому полимеризация таких мономеров -
При T > Tпр и ΔG > 0 полимеризация термодинамически запрещена и возможна лишь деполимеризация макромолекул.
При T = Tпр и в системе устанавливается полимеризацион-но-деполимеризационное равновесие:
kp
∼Mn* + М < ---- >∼Мn+1*
kd
kp[Mn*][Mр]=kd[Mn+1*],
где [Mp] - равновесная концентрация мономера.

Слайд 21где ΔGо = - RTlnKp - стандартный потенциал при Р =
Величины - называются стандартными и относятся к превращению чистого мономера (или одного моля мономера в растворе) в аморфный полимер

Слайд 22Кинетические параметры
Кинетические параметры позволяют описать скорость реакции, а также определить
Способность мономеров к полимеризации зависит от этих параметров. Если термодинамически реакция не разрешена, то ее нельзя провести ни при каких условиях. Однако, если термодинамика разрешает проведение процесса, то это не значит, что он может быть проведен в любых условиях. В этом случае определяющую роль играют кинетические параметры.

Слайд 23Длина цепи
Полимеризация вызывается первичными активными центрами, образующимися из специально вводимых соединений
- инициаторов при радикальной полимеризации,
катализаторов при каталитической,
или в результате физического воздействия на систему.
Для цепных реакций существует понятие длины кинетической цепи, равной числу химических актов, возбужденных одной исходной активной частицей, например радикалом.
При полимеризации развитие кинетической цепи, сопровождается ростом материальной цепи - макромолекулы.
В зависимости от механизма полимеризации длина кинетической цепи может быть равна длине материальной цепи, но может быть больше или меньше ее.

Слайд 24Элементарные стадии процесса полимеризации
Полимеризационные процессы в общем случае протекают в три
- зарождение цепи,
- рост полимерной цепи,
-обрыв полимерной цепи
Процессы полимеризации могут протекать как в присутствии инициаторов, так и под воздействием разнообразных каталитических систем.
Различие между ними заключается в том, что распад инициаторов приводит к образованию первичных активных центров и они в виде осколков входят в полимерную цепь.
Катализаторы ускоряют процесс полимеризации (снижают энергетический барьер реакции), но в состав цепи не входят.

Слайд 25Методы проведения процесса
Полимеризация мономера может проводиться в растворителе и в отсутствие
Если образующийся полимер растворим в мономере и растворителе, то полимеризация протекает без разделения фаз и называется гомогенной.
Если они несовместимы, то образующийся полимер выделяется в отдельную фазу, и такая полимеризация называется гетерофазной.
Внешним признаком гетерофазной полимеризации является помутнение полимеризующейся системы.

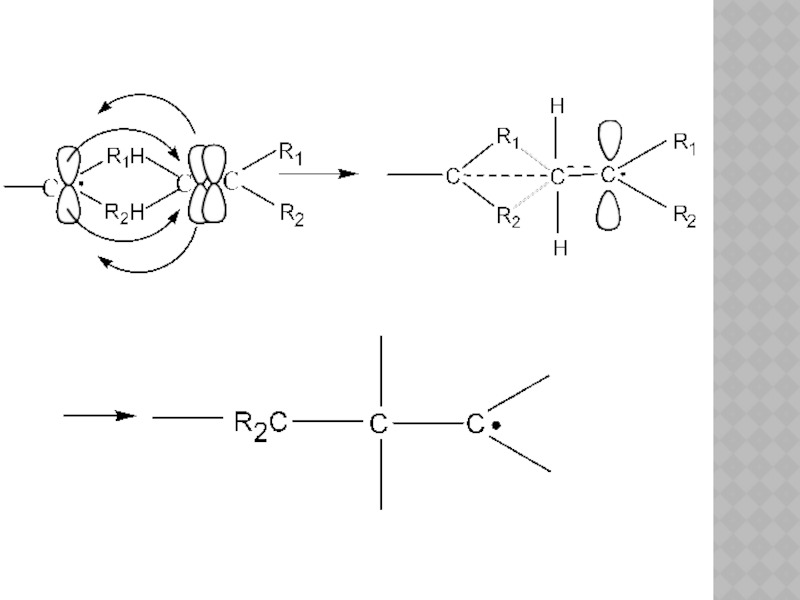
Слайд 26Радикальная полимеризация
Радикальная полимеризация – полимеризация, инициируемая свободными радикалами, генерируемыми в реакционной
В этом случае в качестве активного центра выступает свободный радикал.
По радикальному механизму полимеризуется большинство мономеров, которые имеют двойную связь С=С.

Слайд 28Мономеры, способные вступать в реакцию радикальной полимеризации
Легко полимеризующимися производными этилена являются:
-
- мономеры, имеющие неявную систему сопряжения двойных связей,
- мономеры, имеющие систему сопряжения двойной связи с неподеленной парой электронов
СН2=СН---СН=СН2
1 2 3 4
Активность мономеров в процессе радикальной полимеризации зависит от их природы и строения.

Слайд 29Введение заместителей в молекулу бутадиена приводит к изменению ее активности в
Введение малополярных заместителей (типа СН3) в положение 2 приводит к повышению активности мономера в радикальной полимеризации. Аналогичный эффект наблюдается при введении малополярного заместителя в положение 3. В то же время введение таких заместителей в положение 1 или 4 приводит к снижению скорости радикальной полимеризации.
Введение полярных заместителей даже в положение 1 или 4 увеличивает скорость реакции. Эффект более значителен, если полярный заместитель вводится в положение 2 или 3.


Слайд 31Мономеры с неявной системой сопряжения двойных связей
стирол,
α-метилстирол,
нитрил акривой кислоты, CH2=CH-C≡N (НАК),
акролеин
Мономеры, имеющие сопряжение двойной связи с неподеленной парой электронов
хлористый винил СН2=СНСl
СН2=СН
:Сl:
хлористый винилен Н2=С(Cl)2
метилвиниловый эфир СН2=СН-О-СН3
,акролеин СН2=СН-С(=О)НМономеры, имеющие сопряжение двойной")
Слайд 32Механизм радикальной полимеризации
Радикальная полимеризация инициируется свободными радикалами - активными частицами, имеющими
Свободные радикалы могут возникнуть в результате действия на систему физических факторов, а также чисто химическим путем - при гомолитическом распаде соединений с относительно невысокими энергиями связи или в результате протекания окислительно-восстановительных процессов.
Радикальная полимеризация является цепным процессом. И как всякий цепной процесс протекает через три этапа.
Элементарные реакции радикальной полимеризации
Инициирование
Рост цепи
Обрыв цепи
Передача цепи
Ингибирование

Слайд 33Инициирование
Инициирование - образование активных центров в виде свободных радикалов
Свободный радикал -
H
RH2C. R:C.
H
Возможные пути инициирования
Свободные радикалы могут возникнуть в результате действия на систему физических факторов, а также чисто химическим путем - при гомолитическом распаде соединений (инициаторов) с относительно невысокими энергиями связи или в результате протекания окислительно-восстановительных процессов.

Слайд 34Гомолитический распад активируется под действием тепла, света, различных активных излучений и
Для того, что бы молекула перешла в активное состояние, необходимо, чтобы она приобрела определенное количество избыточной энергии. Избыточная энергия, необходимая для преодоления энергетического барьера реакции называется энергией активации.
Чем выше энергия активации, тем меньшее число молекул в данных условиях являются активными.

Слайд 35Для перевода молекул в активное состояние существуют следующие методы
1 Инициирование под
- термическое инициирование,
- фотохимическое инициирование,
- радиационное инициирование
2. Химическое инициирование - под воздействием специальных веществ – инициаторов.
Инициаторы способны распадаться на свободные радикалы:
- под действием тепла,
- в присутствии окислительно-восстановительных систем.

Слайд 36Инициирование под действием физических факторов
Термическое инициирование
Мономолекулярный механизм инициирования Штаудингера
Бимолекулярный механизм инициирования

Слайд 37Экспериментально установлено, что растущая полимерная цепь - монорадикал, поскольку длинные гибкие
Доказано, что бирадикал на начальных стадиях процесса превращается в монорадикал путем внутримолекулярного переноса цепи:

Слайд 38Фотохимическое инициирование
Инициирование связано с поглощением молекулой мономера кванта световой энергии, энергия
Е = hν
При действии УФ-света на мономера могут образоваться свободные радикалы:
Если мономер не поглощает свет в используемой области, то необходимо использование фотосенсибилизатор (соединение с развитой системой со-пряжения, например, бензофенон С6Н5СОС6Н5).
В некоторых случаях добавка может действовать как фотоинициатор (сама распадается на радикалы, например, перекиси).
Скорость фотоинициируемой полимеризации пропорциональна корню
квадратному из интенсивности облучения и концентрации фотосенсибилизатора.
К процессам радикальной полимеризации на свету способны стирол, ММАК, МВК, МАК.

Слайд 39Радиационно-химическое инициирование
Протекает под воздействием ионизирующих излучений (α, β, γ-излучение). При этом
Молекулы мономера при действии ускоренных электронов или γ-излучения ионизируются, а затем дают радикалы:
Энергия возбуждения может превысить энергию разрыва химической связи, в результате молекула распадается на радикалы. Таким образом можно инициировать привитую полимеризацию.
Термический, фото- и радиационно-химический способы инициирования хотя и обладают рядом преимуществ, сопровождаются различными побочными реакциями (разветвления, деструкции и т.д.), поэтому чаще используют химическое инициирование.
. При этом образуются активные частицы либо")
Слайд 40Химическое инициирование
Химическое инициирование является наиболее распространенным способом перевода молекул в активное
- под действием тепла,
в присутствии окислительно-восстановительных систем.
Инициаторы должны удовлетворять следующим требованиям:
- легкая доступность,
- дешевизна,
- стабильность при комнатной температуре,
- легкий распад при сравнительно невысоких (до 150 оС) температурах с заметной скоростью на свободные радикалы.
Энергия разрыва связей

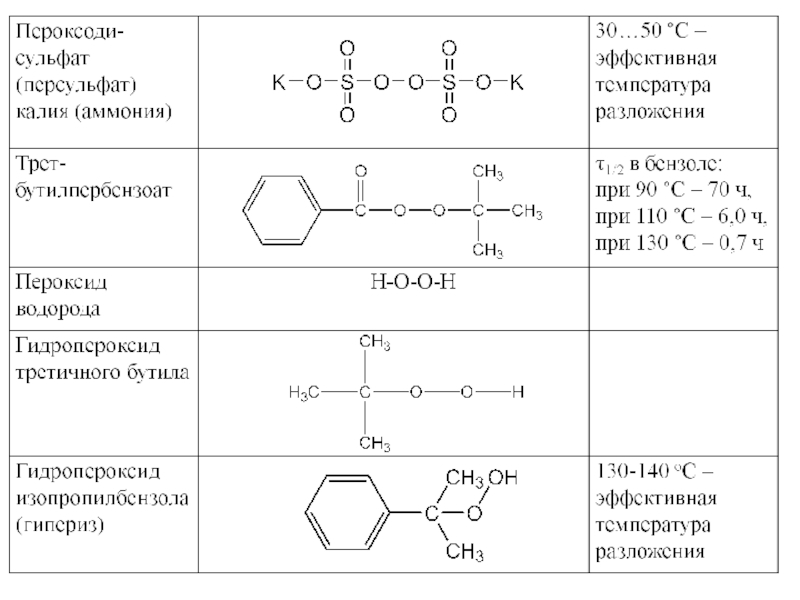
Слайд 41В качестве инициаторов используются следующие классы соединений:
пероксиды ROOR,
гидроперекиси ROOН,
азо- (R1-N=N-R2) и
ди- и полисульфиды,
металлорганические соединения.
Природа образующихся свободных радикалов зависит от типа применяемого инициатора и механизма его разложения.
Наиболее часто применяемые инициатора радикальной полимеризации
 и диазосоединения (RR’CN2), ди- и")
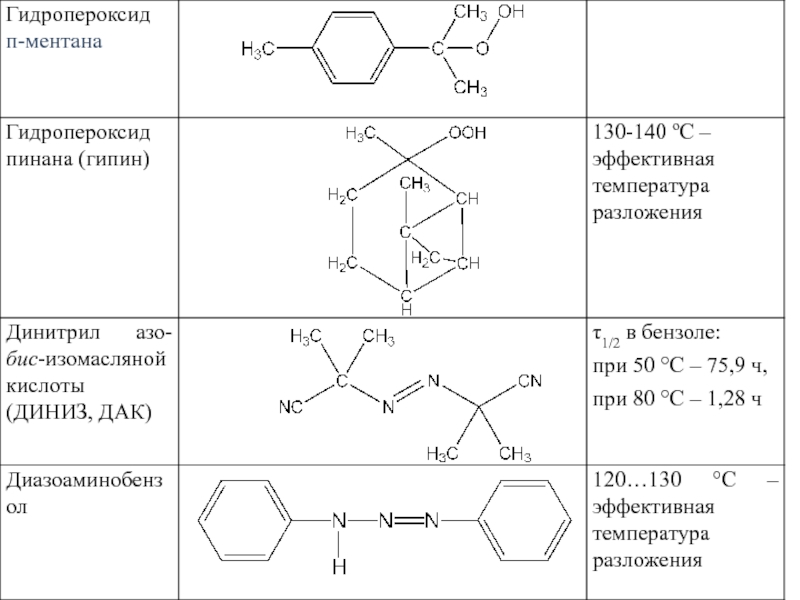
Слайд 44Период полураспада (τ1/2):
Распад пероксида бензоила
Распад третичных гидропероксидов
Гидропероксид третичного бутила
: Распад пероксида бензоила Распад третичных гидропероксидовГидропероксид третичного бутила может распадаться, давая органический")
Слайд 45
Распад динитрила азо(бис)изомасляной кислоты
Распад диазобензола
С6Н5-N=N-NH-С6Н5 N2 + (С6Н5)∙ + (С6Н5NH∙)
Эффективность инициирования (f) - отношение количества осколков инициатора, вошедших в состав макромолекулы, к общему количеству свободных радикалов, образовавшихся в данных условиях.
изомасляной кислоты Распад диазобензола")
Слайд 46Эффект клетки (эффект Франко-Рабиновича)
Уменьшение количества свободных радикалов в жидкой фазе по
При распаде инициатора в газовой фазе свободные радикалы разлетаются на расстояние свободного пробега.
В жидкой фазе вследствие высокой вязкости среды скорость разбегания значительно ниже.
Свободные радикалы существуют некоторое время в окружении молекул мономера, растворителя (как бы находятся в клетке) и этого времени достаточно для того, что бы прошел процесс их рекомбинации.
Разделение радикальной пары протекает:
- за счет диффузии,
за счет взаимодействия свободного радикала с молекулой мономера (или с молекулой среды).
1. ROOR [RO∙…О∙R]
2. Если радикалы не успевают выйти из клетки, то происходит обратный процесс рекомбинации за счет обратимости процесса
[RO∙…O∙R] ROOR
Уменьшение количества свободных радикалов в жидкой фазе по сравнению с газовой фазой.")
Слайд 473. Выход из клетки:
- за счет диффузии
[RO∙…O∙R]
- за счет взаимодействия с молекулой мономера или растворителя (способ эстафетной передачи)
[RO∙…O∙R] + М1 ROМ1∙ + O∙R
Полимеризация в массе. Увеличение выхода полимера сопровождается ростом вязкости системы, что затрудняет выход радикалов из клетки. При этом выход из клетки происходит за счет взаимодействия радикала с молекулой полимера, т.е. происходит передача цепи на полимер. Поэтому при полимеризации в массе обычно происходит образование разветвленных и сшитых полимеров.
Основной недостаток большинства химических инициаторов заключается в необходимости проведения процесса полимеризации при повышенных температурах. Даже наиболее активные инициаторы промышленного типа – персульфаты калия и аммония – позволяют проводить процесс при температуре около 40…50 °С (таблица 7). Между тем при снижении температуры полимеризации существенно улучшаются технические свойства многих полимеров.

Слайд 48Окислительно-восстановительное инициирование
1 При введении в реакционную систему, наряду с основным инициатором
2 В присутствии окислительно-восстановительных систем (ОВС) эффект клетки отсутствует, поскольку распад инициатора протекает с образованием одного радикала.
3 ОВС обладают более низкой энергией активации образования радикалов (~40 кДж/моль) в сравнении с обычными инициаторами (~120…160 кДж/моль). Поэтому ОВС можно применять в широком интервале температур.
Окислительно-восстановительные системы делятся на три типа:
1. Системы, приводящие к образованию одиночного радикала.
2. Системы, приводящие к образованию 2 R∙ (Гидрохинон-Сульфидная).
3. Системы, приводящие к образованию промежуточных соединений, которые быстро распадаются на свободные радикалы.

Слайд 49Системы, приводящие к образованию одиночного радикала
- Инициирующая система пероксид водорода –
H-O-O-H + Fe2+ → НO⋅ + HO¯ + Fе3+
- Инициирующие системы на основе органических окислительно-восстановительных систем (пероксид бензоила – диметиланилин):

Слайд 50
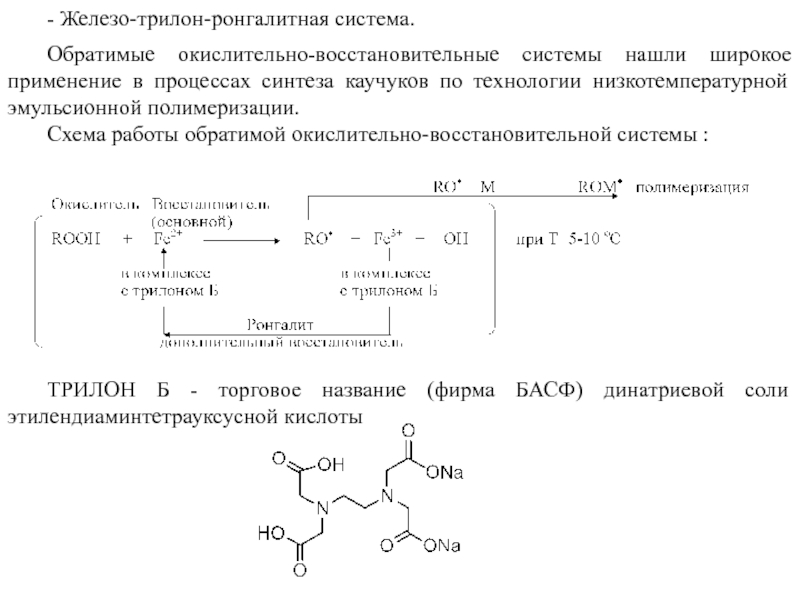
- Железо-трилон-ронгалитная система.
Обратимые окислительно-восстановительные системы нашли широкое применение в процессах синтеза каучуков по технологии низкотемпературной эмульсионной полимеризации.
Схема работы обратимой окислительно-восстановительной системы :
ТРИЛОН Б - торговое название (фирма БАСФ) динатриевой соли этилендиаминтетрауксусной кислоты
Слайд 51В данном рецепте Fe2+ может использоваться в виде пирофосфатных или этилендиаминотетраацетатных
Структура комплекса с динатриевой солью этилендиаминтетрауксусной кислоты :
Наличие координационных связей между атомами железа и азота приводит к снижению степени диссоциации соли, что обеспечивает постепенное и равномерное расходование ионов Fe2+.
В качестве дополнительного восстановителя применяют ронгалит, при окислении которого ионами Fe3+ происходит регенерация ионов Fe2+.
→ 2Fe2+ +
ронгалит
(натриевая соль формальдегидсульфоксиловой кислоты)

Слайд 52Первичные свободные радикалы, образовавшиеся по любому из выше перечисленных способов, далее
Стадия роста полимерной цепи
R∙ + M1 R M1∙
R M1∙+ M2 R M1M2∙
…………………………………
R Mn-1∙+ Mn R Mn∙
Стадия роста цепи характеризуется малой энергией активации (12÷40 кДж/моль) и большой скоростью реакции (для большинства мономеров kр = 102÷104 л/моль⋅с).
Константы скорости и энергия активации роста цепи зависят от природы мономера и параметров реакционной среды.

Слайд 53В ходе реакции происходит выделение тепла.
кДж/моль
Образование σ = 332,3 кДж/моль
Разрыв π-связи
332,3 - 256,5 = 75,8 ккал/моль
При радикальной полимеризации обычно образуются полимеры, имеющие атактическую структуру.
В этом процессе практически нельзя получить стереорегулярные полимеры.
Атактические полимеры - это полимеры, содержащие звенья всех структурных видов, возможные для данного мономера.
Например, диены могут входить в макромолекулу как звенья в положении 1,4 и 1,2 (1.3-бутадиен). Для изопрена и хлоропрена возможно так же присоединение в положение 3,4. При этом присоединение мономерных звеньев может идти различным путем: голова к голове, голова к хвосту, хвост к хвосту. При радикальной полимеризации получаются статистические полимеры.

Слайд 54
Стадия обрыва полимерной цепи
Возможные пути обрыва цепи
1. Рекомбинация (Основной)
Взаимодействие растущего полимерного
~
~
~
~
~
неактивная полимерная
цепь (основной случай)
2. Диспропорционирование
Реакция перераспределения водорода - заключается в передаче атома водорода от одного свободного радикала к другому свободному радикалу. В результате образуется две неактивные полимерные цепи, из которых одна имеет на конце двойную связь.
~
~
~
~
Взаимодействие растущего полимерного радикала с любым другим")
Слайд 55
3. Столкновении свободных полимерных радикалов с примесями, находящимися в реакционной среде.
В
RM∙n + X RMnX ∙
4. Реакции передачи или переноса цепи
RM∙n + XН RMnН + X ∙
агент новый активный
передачи цепи центр
X ∙ + М1 XМ∙1
XМ∙1 + М2 XМ1М∙2
Таким образом, в присутствии агента передачи цепи происходит образование нового свободного радикала.

Слайд 56
При этом могут возникнуть следующие ситуации:
а) активность нового свободного радикала может
б) активность нового свободного радикала может быть меньше активности предшествующего радикала. При этом наблюдается снижение как скорости, так и ММ.
в) активность нового свободного радикала может быть значительно меньше активности предшествующего радикала. В этом случае агент передачи цепи может выступать замедлителем или ингибитором процесса радикальной полимеризации.
Агентами передачи цепи могут выступать:
растворитель,
мономеры,
специальные агенты передачи цепи,
инициатор,
полимер,
другие примеси.
 активность нового свободного радикала может быть равна активности предшествующего")
Слайд 57
Способность к участию в передаче цепи при полимеризации характеризуют константами передачи
Передача цепи на растворитель
Эффективными агентами передачи цепи при радикальной полимеризации являются хлорированные углеводороды: СCl4, CHCl3.
Наиболее высокое значения СS = 9⋅10-3 имеет СCl4. Наиболее инертным растворителем является бензол СS=10-5

Слайд 58
Передача цепи на мономер
~
~
Для большинства мономеров значение См невелико (0.1÷5)⋅10-4.
Однако
Процессы, в результате которых получаются низкомолекулярные полимеры, называются процессами теломеризации.
Передача цепи на агент передачи цепи
Чаще всего в процессах радикальной полимеризации применяют специальные агенты передачи цепи, добавка которых в рецепт (в небольших количествах) приводит к резкому снижению ММ. Такие вещества называют регуляторами длины молекулярной цепи. В качестве таких регуляторов применяют
тр-ДДМ (тр-С12Н25SH),
н-BuSH (СS=2.2),
⋅10-4. Однако при полимеризации мономеров, содержащих")
Слайд 59
Бис(изопропилксантоген)дисульфид, дипроксид
CBr4 (СS=1.4)
Дипроксид, как регулятор ММ, является более активным, чем
Трет-ДДМ расходуется постепенно и обычно сохраняется до конца процесса полимеризации. Как следствие, ММ полимера при увеличении его выхода растет в меньшей степени. Поэтому трет-ДДМ используют чаще, а при применении Дипроксида используют дробную подачу (в два приема).
дисульфид, дипроксид CBr4 (СS=1.4)Дипроксид, как регулятор ММ, является более активным, чем трет-ДДМ. В то же")
Слайд 60
Передача цепи на инициатор
Различные инициаторы обладают различной константой передачи цепи.
Азонитрилы
Многие пероксиды характеризуются весьма значительными константами передачи цепи.
Среди инициаторов наиболее сильными агентами передачи цепи являются гидропероксидные соединения.
При получении низкомолекулярных полимеров иногда пользуются этим методом и увеличивают количество инициатора в десятки раз (обычно в 5).

Слайд 61
Передача цепи на полимер
В том случае, когда мономера в реакционной среде
1/Pn = 1/Po + ks[S]/[M]
где Po - среднечисловая степень полимеризации полимера, полученного в отсутствие агента передачи цепи,
Pn - среднечисловая степень полимеризации полимера, полученного в присутствии агента передачи цепи,
ks - константа передачи цепи,
[S] - концентрация агента передачи цепи,
[M] - концентрация мономера.
Определение ks
Синтезируют набор полимеров, полученных при различных значениях [S]. Определяют Pn для всех полимеров и строят зависимость
1/Pn = f([S]/[M])

Слайд 62
1/Pо
[S]/[M]
tgϕ = ks
ks оценивают для полимеров, полученных при низких степенях превращения мономера. В этом случае концентрация полимера низка и ею пренебрегают.

Слайд 63
Реакции замедления и ингибирования процесса радикальной полимеризации
В зависимости от того, какое
1 - кинетическая кривая, полученная при проведении процесса в отсутствие ингибиторов и замедлителей,
2 - хинон = 0,005 %, задержка 2 часа,
3 - хинон = 0,01 %, задержка 4 часа.
4 - кинетическая кривая, полученная при проведении процесса в присутствии замедлителя,
5 - кинетическая кривая, полученная при проведении процесса в присутствии замедлителя и ингибитора.

Слайд 64
Время задержки процесса полимеризации называют индукционным периодом. Индукционный период зависит от
Гидрохинон и п-фенилендиамин ингибируют процесс полимеризации лишь в присутствии кислорода, поскольку истинными ингибиторами являются соединения, имеющие хиноидную структуру
В качестве замедлителей могут использоваться О2, I2, Нафтам-2 (N-фенил-2-нафталамин) и др. Механизм действия замедлителей и ингибиторов одинаков, разница только в силе их действия.

Слайд 65
Механизм действия замедлителей и ингибиторов
Важным фактором является то обстоятельство, что многие
п-хинондиимин

Слайд 66
Образующийся хинон взаимодействует со свободными радикалами, давая семихиноидный радикал (бензоидная структура).
Переход
Константа Си = 518 для бензохинона при полимеризации стирола при 50°С.
Хиноны являются своеобразными ловушками для свободных радикалов, поскольку выводят свободные радикалы из зоны реакции. Однако сами при этом так же расходуются. В связи с этим индукционный период пропорционален количеству ингибитора.
.Переход в бензоидную структуру является")
Слайд 67
Замедлители по своему действию отличаются от ингибиторов тем, что скорость их
N-Фенил-β-нафтиламин
(Неозон Д)

Слайд 68
Роль кислорода в процессах радикальной полимеризации
Кислород может выполнять две роли:
При Т
При Т < 50 оС кислород выступает ингибитором процесса радикальной полимеризации
1 Полимеризация ММА в вакууме при 100 оС
Примеры
2 Полимеризация этилена. Т =200 оС, Р=150МПа.
Метилметакрилат (ММА) — Сложный метиловый эфир метакриловой кислоты

Слайд 69
Реакции разветвления и перекрещивания цепей
Реакции разветвления и перекрещивания цепей обычно называют
Образующиеся структуры значительно ухудшают свойства получаемых полимеров.
Такие полимеры плохо обрабатываются, становятся нерастворимыми.
В связи с этим при проведении радикальной полимеризации необходимо стремиться к получению полимеров линейной структуры.
Поэтому радикальную полимеризацию обычно прекращают при выходе полимера 65-70 % (для каучуков). Замечено, что с понижением температуры реакции вероятность побочных реакций значительно снижается.
Это связано с тем, что энергия активации побочных реакций значительно выше, чем энергия активации реакции роста полимерных цепей в линейном направлении.
Следовательно, чем ниже температура, тем больше вероятность получения полимеров линейной структуры.
 реакциями. Образующиеся")
Слайд 70
Некоторые особенности полимеризации диенов
При полимеризации диенов в зависимости от условий проведения
димеры циклического строения (Т > 50 оС),
линейные и разветвленные димеры и тримеры (Т > 50 оС),
сшитые, трехмерные полимеры (Т > 50 оС).
Данное обстоятельство связано с различием энергий активации процессов димеризации и полимеризации (первая выше второй).
Димеры диенов (дивинила) имеют циклическое строение
Димеры дивинила такого строения неспособны к радикальной полимеризации, поэтому нежелательны.

Слайд 71
Аутополимеризация
(самопроизвольная полимеризация)
Аутополимеризация - процесс самопроизвольной полимеризации, протекающий без специального внешнего
Такое явление обычно наблюдается при хранении мономеров, не заправленных ингибиторами, и в присутствии следов кислорода.
Такие полимеры не растворяются и даже не набухают. Они не вулканизуются и технически не ценны.
Кроме того, образование таких полимеров опасно, т.к. даже небольшие количества такого полимера быстро увеличиваются в объеме. Они могут образовываться в аппаратах или арматуре, и их образование представляет большую опасность, так как может привести к забивке или разрыву стенок сосудов и трубопроводов.
Для ингибиторов полимеризации Си >> 1 (предотвращают самопроизвольную полимеризацию мономера при хранении). Ингибиторы могут присоединяться к радикалу с образованием стабильного радикала, неспособного продолжать кинетическую цепь, или насыщенного соединения (если ингибитор - стабильный радикал).
Аутополимеризация - процесс самопроизвольной полимеризации, протекающий без специального внешнего воздействия. При этом образуются")
Слайд 72
Передача цепи на полимер
Кроме всего прочего возможна передача цепи на полимер,
При этом увеличивается разветвленность макромолекул:
где ρ - плотность ветвления (число ветвлений на одно элементарное звено), Ср - константа передачи на полимер, х - степень конверсии.
Для уменьшения влияния ветвления (возможны сшивки!) вводят регуляторы или проводят процесс полимеризации гетерофазно.

Слайд 73Гель-эффект (эффект Тромсдорфа)
Время жизни растущих макрорадикалов мало (10-6-10-9 с), однако при
Например, при полимеризации в массе при 15-25% превращения мономера в полимер вязкость увеличивается в 10 000 - 100 000 раз.
С повышением вязкости системы диффузия клубка в клубок замедляется и, начиная с некоторой глубины полимеризации, лимитирует обрыв цепи. Вследствие этого число активных центров растет и скорость полимеризации увеличивается.
Ускорение полимеризации на глубоких стадиях, вызванное увеличением вязкости системы и диффузионными затруднениями стадии обрыва цепи и носит название Гель-эффект или эффекта Тромсдорфа.
Гель-эффект уширяет ММР до коэффициента полидисперсности 5-10. Часто справедливо равенство kо⋅η=const, т.е. чем выше вязкость, тем меньше вероятность обрыва цепи.
Время жизни растущих макрорадикалов мало (10-6-10-9 с), однако при снижении температуры и увеличении")
Слайд 74
Кинетический анализ реакции инициированной радикальной полимеризации
На кинетике реакции полимеризации и свойствах
Общее кинетическое уравнение полимеризации может быть выведено без учета реакций передачи цепи. Если допустить, что активность макрорадикала не зависит от его длины и мономер расходуется только на стадиях инициирования и роста цепи, то скорость полимеризации (или скорость расходования мономера) можно описать уравнением:
Где - скорости инициирования и роста.

Слайд 75
Концентрацию радикалов, которую трудно измерить, можно исключить из рассмотрения, используя принцип
Тогда для стационарного участка процесса полимеризации:
Из уравнения следует, что скорость полимеризации увеличится в
раз при удвоении скорости инициирования, это объясняется бимолекулярностью реакций обрыва. Пропорциональность скорости полимеризации от концентрации мономера выполняется не всегда и, как правило, степень больше 1, что связано с участием мономера на стадии инициирования и в реакциях передачи цепи.

Слайд 76
Длина кинетической цепи ν (т.е. количество присоединившихся молекул мономера, приходящееся на
Средняя степень полимеризации при рекомбинационном обрыве цепи:
при диспропорционировании:
Среднечисленная степень полимеризации
")
Слайд 77
Во многих случаях встречаются оба эти два варианта обрыва цепи:
где λ
Т.е. увеличение концентрации радикалов и, следовательно, скорости радикальной полимеризации приводит к образованию макромолекул меньшей молекулярной массы.
Длина кинетической цепи при Т=const определяется природой мономера и не зависит от способа инициирования, а также от природы инициатора.

Слайд 78
С учетом реакций передачи цепи:
где
Выражая концентрацию радикала через скорость полимеризации
и используя константы передачи на мономер и растворитель, получаем:
Если проводить полимеризацию в присутствии разных количеств инициатора и определить значения и , то, используя (как правило
линейную) зависимость , можно определить значения
константы СМ, а также отношение констант обрыва и роста.

Слайд 79
Например, отношение
различных мономеров при [M]=const, [I]=const при 60°С для полимеризующегося с высокой скоростью винилацетата равна 0.33, а для стирола 0.022.
Молекулярно-массовое распределение
При радикальной полимеризации ММР обычно получается широким, а конкретный тип распределения в условиях гомогенного процесса определяется механизмом ограничения растущих цепей:
в случае диспропорционирования и (или) передачи Mw /Mn ≈ 2 (распределение ФЛОРИ), а при обрыве рекомбинацией Mw /Mn ≈ 1.5 (распределение Шульца).
При параллельном осуществлении обеих реакций бимолекулярного обрыва - это отношение имеет промежуточное значение.
При полимеризации до глубоких степеней превращения мономеров или при образовании нерастворимого полимера наблюдается значительное уширение ММР, вплоть до появления полимодального ММР.

Слайд 80
Влияние различных факторов
на процесс радикальной полимеризации
Влияние температуры
Скорость реакции радикальной полимеризации
Температурный коэффициент большинства химических реакций близок к 2, т.е. применимо правило Вант-Гоффа:
"повышение температуры на 10о приводит к увеличению скорости реакции приблизительно в два раза".
где kТ2 и kТ1 - константы скорости реакции при температурах t2-t1, соответственно.
Следствие: чем больше энергия активации, тем большее влияние оказывает изменение температуры на скорость радикальной полимеризации.

Слайд 81
Энергии активации и константы скорости роста
цепи для некоторых мономеров
Увеличение скорости полимеризации
V2 = k2 [M∙n] [M]
Но одновременно с увеличением скорости расходования мономера увеличивается и скорость обрыва цепи.
V3 = k3 [M∙n]2
Учитывая значения показателя степени у [M∙n], скорость обрыва цепи увеличивается во много раз больше, чем скорость роста полимерной цепи. Следствие: при повышении температуры наблюдается значительное снижение ММ полимера

Слайд 82
Влияние инициатора
С увеличением концентрации инициатора [I] увеличивается число свободных радикалов [R∙].
- скорость реакции возрастает (за счет увеличения числа активных центров).
- степень полимеризации и ММ уменьшаются.
Влияние давления
Повышение давления до нескольких МПа практически не оказывает влияния на процесс радикальной полимеризации.
Однако значительное повышение давления до 300-500 МПа приводит к увеличению скорости процесса радикальной полимеризации.
Особенностью поведения процесса при высоких давлениях является тот факт, что оно практически не влияет на ММ.

Слайд 83
Способы проведения радикальной полимеризации
Радикальную полимеризацию проводят в основном:
- в блоке (массе),
- растворе,
- эмульсии,
- суспензии,
- газовой фазе.
При этом процесс может протекать в гомогенных или гетерогенных условиях.
Кроме того, фазовое состояние исходной реакционной смеси может также меняться в ходе полимеризации.
Полимеризация в блоке (в массе)
«+»: Процесс проводится в отсутствие растворителя, благодаря чему не происходит загрязнения полимера, возможность получения полимера в форме сосуда, в котором проводится процесс без какой-либо дополнительной обработки.
«―»: Процесс трудно поддается регулированию вследствие высокой экзотермичности полимеризации. Рост вязкости среды, что затрудняет отвод тепла и приводит к местным перегревам и деструкции полимера, неоднородности его по молекулярной массе.
, - растворе, - эмульсии,")
Слайд 84
Полимеризация в растворе
Отсутствуют многие недостатки блочной полимеризации:
- устраняется возможность местных
- уменьшается вязкость реакционной системы, что облегчает ее перемешивание.
Недостатки:
- в ряде растворителей возрастает доля реакций передачи цепи, что приводит к уменьшению молекулярной массы полимера,
- полимер может быть загрязнен остатками растворителя, который не всегда легко удаляется из полимера.
Полимеризацию в растворе проводят двумя способами:
- для полимеризации применяют растворитель, в котором растворяется и мономер, и полимер. Получаемый раствор используют как таковой или выделяют полимер осаждением либо испарением растворителя.
- полимеризацию проводят в жидкости, в которой растворяется мономер, но не растворяется полимер. Полимер по мере образования выпадает в твердом виде и быть отделен фильтрованием.

Слайд 85
Полимеризация в суспензии
(Бисерная или гранульная полимеризация) широко используется для синтеза различных
Мономер диспергируют в воде в виде мелких капелек. Устойчивость дисперсии достигается механическим перемешиванием и введением в реакционную систему специальных добавок - стабилизаторов.
При полимеризации в суспензии применяют растворимые в мономере инициаторы. Процесс осуществляется в каплях мономера, которые можно рассматривать как микрореакторы блочной полимеризации.
Достоинство - хороший отвод тепла.
Недостаток - возможность загрязнения полимера остатками стабилизатора.
Газофазная полимеризация
Мономер (например, этилен) находится в газообразном состоянии. В качестве инициаторов могут использоваться кислород и пероксиды. Процесс протекает при высоком давлении.
 широко используется для синтеза различных полимеров. Мономер диспергируют в")
Слайд 86
Твердофазная полимеризация
Это полимеризация мономеров, находящихся в кристаллическом или стеклообразном состоянии.
Существует два крайних случая перехода мономерного кристалла в полимер (возможно множество промежуточных случаев):
- структура мономерного кристалла существенно определяет структуру полимера, т. н. топотактический процесс (например, полимеризация сопряженных диацетиленов или триоксана).
(Другой пример топотактической полимеризации - радиационно-химическая полимеризация 2,3-диметилбутадиена-1,3 в гексагональных кристаллах мочевины, в которых образуются каналы, заполненные линейными последовательностями мономеров, причем полимер получается стереорегулярным.);
- полимер возникает как самостоятельная фаза в протяженных дефектах кристаллической решетки мономера, что приводит к дальнейшей ломке мономерного кристалла; образующаяся полимерная фаза аморфна (например, полимеризация акриламида).

Слайд 87
Полимеризация в эмульсии
(Латексная полимеризация - ЛП). Латекс - водная коллоидная
При эмульсионной полимеризации (ЭП) в качестве дисперсионной среды используют воду, в качестве эмульгатора - различные мыла.
Для инициирования процесса применяют водорастворимые инициаторы, окислительно-восстановительные системы.
Полимеризация может протекать в молекулярном растворе мономера в воде, на поверхности раздела капля мономера ― вода , на поверхности или внутри мицелл мыла, на поверхности или внутри образующихся полимерных частиц, набухших в мономере.
Достоинства ЭП:
- возможность осуществления процесса с большими скоростями с образованием полимера высокой молекулярной массы,
- легкость теплоотвода;
Недостатки:
- необходимость удаления остатков эмульгаторов,
- большое количество сточных вод, требующих специальной очистки.
. Латекс - водная коллоидная дисперсия полимерных частиц размером")
Слайд 88
Эмульсионная радикальная полимеризация
Процесс впервые реализован в Германии в 1938 году,
-
- в СССР 1949 год.
В настоящее время ЭП является одним из основных методов получения синтетических латексов и полимеров.
Методом ЭП обычно получают бутадиенстирольные латексы и полимеры,
(со)полимеры на основе α-метилстирола

Слайд 89
сополимеры хлоропрена и бутадиена-1,3 и т.д.
сополимеры (мет)акриловой кислоты, (мет)акриламида и нитрила
где R -H или -CH3;
акриловой кислоты, (мет)акриламида и нитрила акриловой кислоты общей структурной")
Слайд 90
Достоинства эмульсионной полимеризации:
Возможность осуществления процесса в непрерывном режиме, что обеспечивает
Низкая вязкость реакционной среды (дисперсия полимера в воде), что позволяет более легко отвести тепло реакции.
Возможность получения широкого ассортимента латексов и полимеров за счет использования различных по природе мономеров, осуществления сополимеризации и смесей латексов.
Возможность получения масло и саженаполненных латексов и СК.
Полная автоматизация.
Высокая безопасность процесса.

Слайд 91
Недостатки:
Невозможность регулирования микроструктуры полимеров особенно при полимеризации малополярных мономеров. Такие
Многокомпонентность системы.
Загрязнение полимера и окружающей среды компонентами системы.
Для проведения процесса ЭП обязательно требуется наличие 2-х фаз:
дисперсионная среда (вода)
дисперсная фаза (мономеры.
В рецепт кроме того обязательно должны входить:
инициаторы (водо- или маслорастворимые),
эмульгаторы - соли канифоли (д-резинаты), соли СЖК (С12-С18), сульфаты (алкилсульфанат, сульфанол), полиионоактивные, амфотерные и неионогенные ПАВ и полимерные ПАВ. В качестве дополнительного стабилизатора выступает лейканол.
Регуляторы ММ - т-ДДМ, Дипроксид,
Активирующая группа (ОВС).

Слайд 92
Механизм эмульсионной полимеризации и топология процесса
Топохимия рассматривает место, где протекают элементарные
Образование ВМС при проведении процесса ЭП протекает по цепному радикальному механизму. Наличие водной фазы и коллоидное состояние диспергированного мономера определяют особенности топохимии процесса ЭП. Все преимущества и все недостатки процесса ЭП определяются гетерогенностью системы и возможностью локализации элементарных реакций ЭП в различных зонах.
Процесс может протекать в :
мицеллах,
адсорбционных слоях эмульгатора,
водной фазе,
каплях мономера,
микрокаплях мономера.

Слайд 93
Трудность создания единой теории ЭП обусловливается следующим:
Многофазность эмульсионной системы.
Многообразие
Такое многообразие обусловлено различием в реакционной способности мономеров, участвующих в процессах ЭП, а так же разным характером распределения мономеров по фазам.
Мицеллярная теория (Юрженко-Харкинса)
Эта теория развита и в основном применительно для неполярных и малорастворимых в воде мономеров (бутадиен, изопрен, стирол, α-метилстирол и др.).
В 1950 годы Юрженко и Харкинс высказали предположение о том, что процесс ЭП протекает в мицеллах эмульгатора. Топохимия процесса эмульсионной полимеризации определяется природой и количеством ПАВ и величиной поверхностного натяжения и коллоидной растворимостью углеводородов в растворах, а так же природой инициатора и др.
Анионоактивные и катионоактивные эмульгаторы в воде находятся в форме ионов, молекул и мицелл.

Слайд 94
Минимальная концентрация эмульгатора, при которой наблюдается образование мицелл, называется ККМ.
Молекулярная растворимость
Мицеллы - агрегаты молекул НМС, абсолютно идентичных и связанных между собой вторичными силами (силами Ван-дер-Ваальса).
Макромолекулы - длинные гибкие молекулярные цепи, звенья которых соединены между собою главными валентными связями.
При низких концентрациях эмульгатора образуются в основном сферическое мицеллы. Величина ККМ зависит от ряда факторов. В частности, в присутствии электролитов наблюдается понижение ККМ.

Слайд 95
С17Н35 СООК Сэмульгатора < ККМ
Истинный раствор Коллоидная система
Некоторый свободный объем

Слайд 96
С мицеллообразованием тесно связан процесс солюбилизации - коллоидной растворимости мономеров в
Количество мицеллярно-растворимого мономера сравнительно невелико.
В одной мицелле обычно растворяется до 100 молекул мономера.
Остальной мономер существует в виде капель эмульсии, которые также окружены молекулами эмульгатора. При этом молекула ПАВ разворачивается углеводородной частью к капле, а полярной частью к воде.
Эмульсионная система является наиболее стабильной в том случае, когда степень адсорбционной насыщенности довольно велика.
Таким образом, в начале процесса практически весь эмульгатор находится в виде мицелл, а мономер в основном в виде капель.

Слайд 97
В том случае, когда в эмульсионной системе процесс протекает в присутствии
В случае малорастворимого в воде инициатора, он попадает внутрь мицелл вместе с мономером, где он распадается на свободные радикалы и начинается процесс ЭП.
Реакция начинается не в каждой мицелле, а лишь в одной из 700, поскольку число свободных радикалов, которые образуются при распаде даже очень активных инициаторов, значительно меньше числа мицелл при обычных концентрациях эмульгатора в реакционной среде.
Вероятность протекания процесса в водной фазе в каплях мономера очень низка. Рост полимерного радикала в мицеллах осуществляется за счет мономера, находящегося в мицеллах. В свою очередь молекулы мономера в мицеллах находятся в динамическом равновесии с молекулами мономера, находящимися в водной фазе. В связи с этим концентрация мономера в мицеллах является постоянной до тех пор, пока существуют капли мономера. Они являются как бы резервуаром, откуда мономер расходуется по мере необходимости.

Слайд 98
По ходу полимеризации мицеллы превращаются в ПМЧ, а молекулы мономера первоначально
1/700 в ПМЧ
в адсорбционные слои ПМЧ.
699
Кинетика процесса в соответствии с этой теорией была описана Смитом и Эвертом
V = k2 [I]0,4 [Э]0,6 [M]

Слайд 99
Кинетическая кривая процесса ЭП
0 - индукционный период,
1 - мицеллярный этап,
2 -стационарный
3 - конечный этап.

Слайд 100
На мицеллярном этапе:
существуют мицеллы, ПМЧ и капли мономера,
поверхностное натяжение постоянно,
число частиц и скорость процесса возрастают.

Слайд 101
Стационарный этап:
существуют ПМЧ и капли мономера,
концентрация мономера внутри
поверхностное натяжение возрастает, поскольку ПМЧ постоянно растут в объеме и их поверхность становиться все более и более адсорбционно-ненасыщенной,
число частиц и скорость процесса постоянны.
Конечный этап
на этом этапе весь мономер перешел в ПМЧ, поэтому капель мономера нет,
концентрация мономера внутри частиц падает,
поверхностное натяжение постоянно,
число частиц постоянно, а скорость процесса падает.

Слайд 102
Теория Медведева-Хомяковского
Согласно этой модели объем ПМЧ не является основной зоной реакции.
Адсорбционный слой имеет определенную толщину и объем и считается, что процесс полимеризации начинается в адсорбционных слоях эмульгатора.
Теория микроэмульгирования
Согласно этой теории роль мицелл в образовании ПМЧ не является определяющей.
Основное значение отводится процессам микроэмульгирования, причем определяющим фактором является дробление капель мономера, что приводит к резкому увеличению поверхности раздела мономер - водная фаза.
Перенос молекул мономера к растущим ПМЧ происходит не путем диффузии, а путем непосредственного контакта ПМЧ с каплями мономера или между собой.

Слайд 103
СОПОЛИМЕРИЗАЦИЯ
Сополимеризацией называют совместную полимеризацию двух или более мономеров.
Она широко используется
Наиболее распространена и изучена бинарная сополимеризация.
Для этого случая можно вывести кинетически (или статистически) без конкретизации механизма и природы активных центров уравнение состава сополимера - зависимость между составами сополимера и исходной смеси мономеров (т.к. как правило они не равны).

Слайд 104
Допущения:
- постоянная скорость инициирования;
- реакционная способность активного центра постоянна;
- все стадии
- мономер расходуется только на рост цепи;
- существует стационарное состояние;
- гомофазная полимеризация;
- сополимер получается с Мn ≥ 104.
- степень конверсии мономеров < 5%, когда состав мономерной смеси мало отличается от исходной.
В этом случае можно записать четыре реакции роста цепи:
~
~
~
~
~
~
~
~

Слайд 105
Скорости исчерпания мономеров при сополимеризации равны:
Поделив эти выражения, получаем отношения концентраций

Слайд 106
В стационарном состоянии устанавливаются стационарные концентрации активных центров каждого типа. Условием
откуда
.
Подставив это значение в выражение отношений концентраций мономерных звеньев, после упрощения получим дифференциальное уравнение состава сополимера Майо-Льюиса:
где
и
- константы сополимеризации, или относительные активности мономеров, равные отношению констант скоростей присоединения к радикалам своего и “чужого” мономеров.

Слайд 107
Параметры r1 и r2 для любой пары мономеров определяются только природой
Это уравнение связывает мгновенные (текущие) концентрации мономеров в сополимере и мономерной смеси через величины относительных активностей мономеров.
По уравнению можно найти константы r1 и r2 при сополимеризации до 5-7% глубины превращения мономеров. При этом условии отношение [M1]/[M2] можно считать равным заданному, а мгновенный состав - равным среднему составу сополимера, образованного на начальной стадии, т.е.
Тогда

Слайд 108
Таким образом химический состав сополимера (при малых степенях превращения) зависит от
Вместо абсолютных молярных концентраций удобнее применять относительные молярные доли.
Уравнение состава может быть решено графически или аналитически.
Графическим выражением уравнения состава являются кривые состава сополимера, вид которых определяется константами r1 и r2 :
 зависит от концентраций мономеров и их")
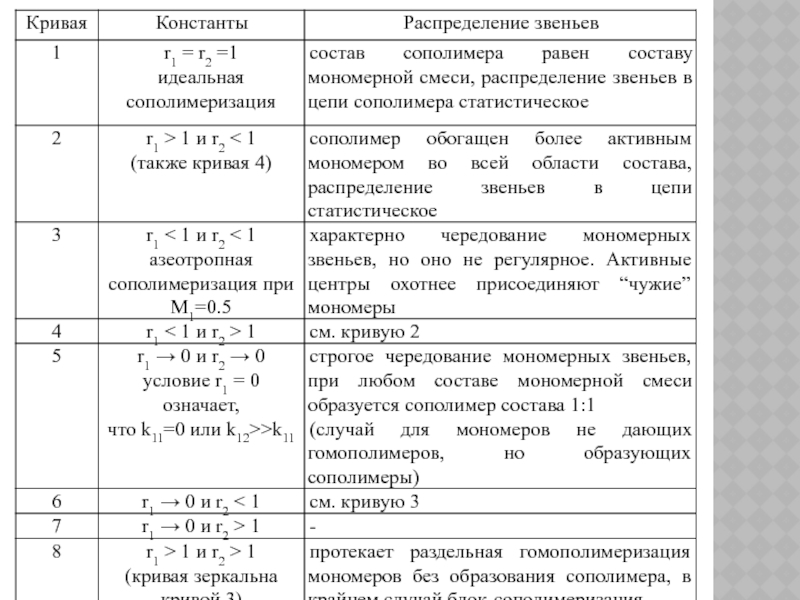
Слайд 110
Статистический анализ чередования звеньев в цепи сополимера указывает на три случая:
при
при r1 r2 < 1 вероятность чередования звеньев увеличивается;
при r1 r2 → 0 в пределе можно регулярно чередующийся сополимер.
Точка пересечения кривой 3 (или кривой 8) с прямой 1 соответствует азеотропной сополимеризации (когда состав сополимера равен составу мономерной смеси).
В промышленных процессах, когда ставится задача получения возможно однородного продукта, руководствуются следующими положениями:
Подбирают мономерные пары с константами сополимеризации, близкими к 1, так чтобы состав мономерной смеси минимально изменялся по ходу реакции;
Реакцию проводят по возможности ближе (или в точке) азеотропного состава;
Ограничиваются минимально возможными конверсиями;
Сохраняют состав мономерной смеси постоянным добавлением по ходу реакции более активного сомономера в реакционную смесь.

Слайд 111
Дифференциальный и интегральный составы сополимеров
Дифференциальный состав сополимера - состав сополимера, образующегося
Интегральный - состав сополимера, образовавшегося к данному моменту времени.
В тех случаях, когда сополимер не имеет характера азеотропа, он будет обогащен одним из мономеров.
При этом более активный мономер будет расходоваться более быстро и в начале процесса сополимеризации сополимер будет обогащен более активным мономером.

Слайд 112
Изменение дифференциального и интегрального составов сополимера в ходе радикальной сополимеризации стирола
Пример
 и бутадиена (75)")
Слайд 113
Константы сополимеризации и их определение
Для определения констант сополимеризации необходимо знать:
состав
состав исходной шихты,
провести не менее 2-х опытов (8-10).
Состав сополимера определяют по данным:
элементного анализа,
приборных физических методов анализа
а) показатель преломления,
б) по плотности,
в) спектральные методы (УФ, ИК, ЯМР).
Если процесс сополимеризации проводить до невысоких конверсии, то можно использовать основное уравнение в дифференциальной форме.
Если процесс проводить более глубоко (боле 15%), то необходимо использовать основное уравнение в интегральной форме.
В настоящее время константы сополимеризации определяют путем решения уравнения сополимеризации при различных степенях превращения мономера с помощью расчетных методов. Такой метод позволяет быстро и с большой точностью рассчитать состав сополимера.

Слайд 114
ИОННАЯ ПОЛИМЕРИЗАЦИЯ
Ионная полимеризация - это цепной процесс, в котором присоединение мономера
Активными центрами на концах цепей являются ионы, образующие с противоионами пару, которые генерируются под действием катализаторов или ионизирующих излучений.
Степень разделения компонентов ионной пары может быть различна. Обычно выделяют три характерных случая:

Слайд 115
Каждая из форм активного центра существенно отличается по активности.
Поэтому для
При изменении диссоциирующей и сольватирующей способности среды процессы ионной полимеризации осложняются различными сопутствующими эффектами (изомеризацией, димеризацией, образованием нестереорегулярного полимера и др.).
Влияние механизма полимеризации стирола на скорость роста цепи (298К)

Слайд 116Внедрение мономера в растущую полимерную цепь осуществляется между заряженным концом растущей
Ионная полимеризация протекает через последовательные элементарные стадии инициирования, роста цепи, передачи и обрыва цепи.
Особенностью ионных полимеризационных процессов является малая вероятность кинетического обрыва цепи, поэтому растущие цепи сохраняют активность и после исчерпания мономера.
Такие цепи называют “живыми”.
Этот эффект используют для получения блок-сополимеров.
Значения энергий активации ионной полимеризации значительно ниже по сравнению с радикальной полимеризацией, поэтому они проводятся при низких температурах, часто отрицательных, так как при понижении температуры увеличивается устойчивость ионых пар.

Слайд 117
Существенный характер имеет выбор растворителя. Нужен растворитель, который обладает хорошей сольватирующей
Наиболее подходящими растворителями, отвечающими этим требованиям, являются: CH3Cl хлористый метил, C5H12 пентан, Ph-NO2 нитробензол и некоторые другие.
В таких растворителях при увеличении сольватирующей способности увеличивается расстояние между ионами в ионной паре, а следовательно изменяется активность центра полимеризации.
Решающее значение имеет химическое строение мономера. Винильные мономеры с электронодонорными заместителями (изобутилен, виниловые эфиры) полимеризуются по катионному механизму, а с электроноакцепторными (акрилонитрил, акрилаты) по анионному. Стирол полимеризуется по различным механизмам, так как за счет сопряжения с кольцом могут стабилизироваться и положительные, и отрицательные, и радикальные центры.
Ионная полимеризация хуже изучена, чем радикальная. Нет единой кинетической схемы и каждый случай описывается своими особенностями кинетики.

Слайд 118
КАТИОННАЯ ПОЛИМЕРИЗАЦИЯ
Впервые катионную полимеризацию осуществил Бутлеров в 1877 году путем полимеризации
Активный центр: растущая цепь имеет положительный заряд - карбокатион.
Мономеры: производные этилена с электронодонорными заместителями, карбонильные гетероциклические соединения, нитрилы. Для ряда мономеров полимеризация возможна лишь по катионному механизму (изобутилен, триоксан, тетрагидрофуран).
Способность винилового мономера к реакциям роста по катионному механизму зависит от степени нуклеофильности двойной связи и возможности образования нуклеофильными группами мономера необратимых комплексов с электрофильными агентами (катионом или кислотой Льюиса).

Слайд 119
Катализаторы:
- протонные кислоты: HClO4, H2SO4, H3PO4, HCl и др.;
- апротонные кислоты
Активны в присутствии сокатализаторов - галогеналкилов, воды, спиртов и других соединений, являющихся донорами ионов водорода.
Излучения высокой энергии и ряд других реагентов: карбениевые Ph3C+, оксониевые R3O+ соли с комплексным противоионом ([SbCl6]−, [PF6]−), алкилпроизводные металлов ZnR2, AlR3, галогенов и межгалогенных соединений: I2, ICl, IBr.
Активность катализаторов может быть разная - например, полимеризация изобутилена в присутствии BF3 происходит со взрывом, с AlBr3 за несколько минут, а с TiCl4 в течение часов.
,")
Слайд 120
Реакция инициирования заключается во взаимодействии катиона с мономером. В простейших, но
HClO4
H+ +СlO4−,
но гораздо чаще в результате диссоциации комплекса катализатора и сокатализатора:
Затем при взаимодействии с мономером катионы смещают на себя электронную плотность π-связи с последующим образованием σ-связи катиона с β-углеродным атомом мономера (по отношению к заместителю):

Слайд 121
Рост цепи в катионной полимеризации происходит путем присоединения мономера к макрокатионам
Скорость роста зависит от степени разделения ионов в ионной паре. Наиболее активными являются свободные ионы. Поэтому увеличение полярности растворителя, как правило, приводит к возрастанию скорости катионной полимеризации, из-за увеличения степени диссоциации ионной пары.
Поскольку реакция диссоциации ионной пары обычно экзотермическая, часто энергия активации процесса катионной полимеризации отрицательна. Т.е. понижение температуры приводит к увеличению скорости катионной полимеризации.

Слайд 122
Обрыв цепи чаще всего происходит в результате мономолекулярной реакции дезактивации активного
~
При этом возможна также рекомбинация ионной пары:
~
Передача цепи на мономер играет исключительную роль в катионной полимеризации, поскольку она определяет предельные значения молекулярной массы полимеров. Эта реакция протекает в результате межмолекулярной передачи катиона водорода:
~
~

Слайд 123
Энергия активации этого процесса положительна, поэтому для увеличения молекулярной массы полимера
Кинетика катионной полимеризации
В большинстве случаев скорость катионной полимеризации пропорциональна концентрациям мономера и катализатора. Однако в результате существования различных форм активных центров, отличающихся по активности (разница в скоростях примерно в 106 раз), часто зависимость скорости процесса имеет более сложный вид.
Соотношение различных типов катионноактивных центров зависит от диэлектрической проницаемости среды ε, основности компонентов и наличия примесей, способных взаимодействовать с катализатором.
Например, в среде CCl4 активные центры более чем на 95% имеют ковалентную форму, а в CH3NO2 - ионную.
Другой пример: полимеризация стирола в присутствии HClO4 при 25°С в смеси дихлорэтана и четыреххлористого углерода ускоряется в 10 000 раз при изменении ε в пределах 2.3-9.7 (путем изменения соотношения растворителей).

Слайд 124
Иногда в процессе полимеризации происходит заметное изменение соотношения между активными центрами,
Уравнение баланса активных центров в момент времени τ для этой системы имеет вид:
где
- общая концентрация активных центров,
- концентра-ция несольватированных центров, kM, kп, kS - константы скорости комплексообразования центров с мономером, полимером и растворителем.
При росте цепи преимущественно на центрах, сольватированных мономером, уравнение скорости полимеризации можно записать в виде:

Слайд 125
Отсюда следует, что константа скорости роста цепи kр зависит от концентрации
Промышленно катионная полимеризация используется для получения полиизобутилена, бутилкаучука, поливинилизобутилового эфира, сополимеров этиленоксида, а также большого числа разнообразных олигомерных продуктов.

Слайд 126
АНИОННАЯ ПОЛИМЕРИЗАЦИЯ
Систематическое изучение анионной полимеризации ненасыщенных соединений началось в 1920-х годах
Активный центр при анионной полимеризации несет частичный или полный отрицательный заряд.
Мономеры, склонные к анионной полимеризации, имеют пониженную электронную плотность С=С-связи электроноакцепторным заместителем (акрилаты, акрилонитрил, этиленоксид, альдегиды, лактоны, лактамы, силоксаны) или имеют повышенную энергию сопряжения (стирол, диены). Кроме того, к анионной полимеризации также склонны многие из карбонилсодержащих соединений и гетероциклов, имеющие связи С=С, С=О, C=N и др.
Катализаторами являются сильные основания, основания Льюиса, т.е. доноры электронов - щелочные металлы, производные металлов I и II группы (алкилы, арилы, алкоголяты, амиды). Процессы, развивающиеся с участием переходных металлов, относят обычно к координационно-ионной полимеризации. Кроме того анионная полимеризация может быть вызвана электрическим током и излучениями высокой энергии.

Слайд 127
Реакция инициирования может осуществляться двумя способами:
- по типу кислотно-основного взаимодействия, в
- по типу окисления-восстановления, в результате переноса электрона между молекулами мономера и катализатора; например, при реакции металлов I группы, а также металлорганические соединения элементов I и II групп. Акту инициирования с участием металла предшествует стадия образования комплекса с переносом заряда (КПЗ) между катализатором и мономером:

Слайд 128
или между металлом и ареном:
На второй стадии мономер вытесняет нафталин из
Процессы по типу окисления-восстановления характерны также для электрохимического и радиационно-химического инициирования. В принципе, при таком механизме возможно параллельное развитие анионных и радикальных реакций, однако в реально изученных системах случаи с заметным участием радикальных процессов не обнаружены.

Слайд 129
Особенностью анионной полимеризации неполярных мономеров является ассоциация катализатора и растущих цепей
Активность анионного катализатора - металлалкила MeR находится в прямой зависимости от полярности связи Me-C, а также от растворителя, причем активные центры могут существовать в виде различающихся по реакционной способности и стереоспецифичности ковалентных поляризованных молекул (II), их ассоциатов (I), ионных пар разной степени сольватации (III,IV), свободных ионов (V):
I II III IV V

Слайд 130
Полимеризация неполярных мономеров (стирола, бутадиена, изопрена) в углеводородных растворителях нередко сопровождается
R-Me + nD
R-Me⋅nD.
Присутствие электронодонора в координационной сфере металла приводит к обеднению электронами и ослаблению связи Ме-С. В реакциях, протекающих с разрывом связи Ме-С, это равнозначно повышению активности катализатора. Например, это явление благоприятствует 1,2 (3,4)-присоединению диенов и образованию синдиотактического полиметилметакрилата.
 в углеводородных растворителях нередко сопровождается индукционными эффектами вследствие недостаточной")
Слайд 131
Рост цепи для анионной полимеризации характерен относительной стабильностью активных центров.
Например,
Это позволяет создать условия для исследования механизма анионной полимеризации, а также для решения различных синтетических задач: получение полимеров с заданным ММР, в т. ч. практически монодисперсных; синтеза полимеров и олигомеров с концевыми функциональными группами, способными к дальнейшим превращениям поликонденсационного или полимеризационного типа, а также блок-сополимеров, привитых сополимеров и различных полимеров с регулируемым типом разветвления и др.
Участие противоиона в актах роста цепи обусловливает большие возможности воздействия на микроструктуру полимера, вплоть до образования в некоторых случаях стереорегулярных и оптически активных полимеров.

Слайд 132
В наибольшей степени ориентирующее влияние противоиона проявляется в углеводородной среде, где
Реакции обрыва и передачи цепи характерны для анионной полимеризации мономеров с полярными функциональными группами. Это всегда более сложный процесс, сопровождающийся дезактивацией активных центров при взаимодействии с функциональными группами мономера и полимера. Энергия активации побочных реакций (как и передача цепи на растворитель в случае веществ с подвижным атомом водорода, например, толуола), как правило, выше, чем энергия роста цепи; поэтому понижение температуры способствует обычно подавлению побочных реакций.

Слайд 133
Чаще всего общей реакцией обрыва цепи является перенос гидрид-иона на противоион
~
~
~
~
Кинетика полимеризации
Анионная полимеризация характеризуется большим разнообразием механизмов реакций и кинетических схем. В каждом конкретном случае выбор инициаторов и условий проведения процесса обусловлен необходимостью синтеза полимера определенной структуры и ММР. Скорость анионной полимеризации особенно при умеренных температурах намного выше скорости радикальной полимеризации. Это связано с более высокой действующей концентрацией активных частиц (в пределе она может быть равна исходной концентрации инициатора). Например, для стирола при 30°С порядок абсолютной константы скорости роста цепи (в л/моль⋅с) при переходе от литиевых ассоциатов II до свободных анионов (V) меняется от 10-1 до 105.

Слайд 134
Общая кинетическая картина анионной полимеризации существенно осложнена упомянутой выше множественностью форм
υр= kр [C0][M]
,
где [M0] и [M] - начальная и текущая концентрации мономера, х=1-[M]/[M0] - степень превращения мономера, n - число растущих концов в макромолекуле.

Слайд 135
Часто наблюдаются более сложные зависимости общего вида:
где учитывается вклад различных форм
Обычно порядок реакции по инициатору варьирует от 1 до 0, а порядок по мономеру в большинстве случаев равен 1.

Слайд 136
ИОННО-КООРДИНАЦИОННАЯ ПОЛИМЕРИЗАЦИЯ
Если акту внедрения мономера в растущую полимерную цепь предшествует акт
В 1954 году А.А.Коротков получил из изопрена стереорегулярный каучук, применив в качестве катализатора литийорганические соединения. При полимеризации на литии или литийорганических соединениях стереорегулярный цис-1,4-полиизопрен образуется лишь в углеводородных средах.
Это объясняется координацией мономера на полярном, но недиссоциированном активном центре
, в результате чего мономерное звено принимает конфигурацию, соответствующую цис-1,4-структуре:

Слайд 137
Добавление всего лишь нескольких процентов электронодонорного соединения (эфира, тетрагидрофурана, алкиламина) резко
Электронодонорное соединение поляризует связь
до разделения на ионы:
~
~
 резко меняет микроструктуру полиизопрена, становится")
Слайд 138
В этом случае происходит координация иона Li+ с концевым звеном макроиона,
что приводит к транс-1,4- и 3,4-структуре.

Слайд 139
В 1955 году немецкий химик Карл Циглер (для получения полиэтилена в
В частности Натта с сотрудниками в Милане исследовал методом дифракции рентгеновских лучей полимеры, полученные из пропилена, и обнаружил, что некоторые изученные полимеры, полученные полимеризацией мономера под действием продуктов реакции триалкилалюминия с хлоридом титана (катализатор Циглера) или под действием трехокиси хрома, нанесенной на окись алюминия, имеют значительно более регулярную структуру, чем другие полимеры полипропилена.
Стереорегулярность полимера сильно влияет на его физические свойства. Например, обычный атактический полипропилен - мягкий резиноподобный материал, тогда как изотактическая модификация представляет волокнистый материал, который можно прясть и ткать. Поэтому не удивительно, что Натта и Циглер получили Нобелевскую премию в 1963 году за открытие стереорегулярных полимеров и катализаторов, необходимых для их получения.

Слайд 140
Типичными катализаторами ионо-координационнной полимеризации являются соединения переходных металлов d-групп (IV-VIII группы
,")
Слайд 141
В акте координации мономер выступает в роли донора π-электронов, а переходный
Рост цепи осуществляется путем внедрения мономера по типу “голова к хвосту”, что связано с преодолением относительно низких активационных барьеров, чем при присоединению по другим типам. Акт координации приводит к определенной ориентации молекулы мономера, обеспечивая раскрытие двойной связи и отбор строго определенной конформации мономера при внедрении в полимерную цепь. В этом случае полимерная цепь будет иметь стереорегулярную изотактическую структуру.
Если отбираемые в акте внедрения конформации мономера противоположны и чередуются регулярно, то образуется стереорегулярная синдиотактическая последовательность.
Известны гетерогенные и гомогенные катализаторы Циглера-Натта, на первых в основном получаются изотактические, а на вторых могут быть получены и синдиотактические полимеры.

Слайд 142
Стереоспецифичность каталитических систем типа Циглера-Натта обусловлена влиянием лигандного окружения в координационной
Таким образом, инициирование стереоспецифической полимеризации протекает по трехстадийному механизму - координации, ориентации и внедрения.
В этих процессах интенсивность реакций ограничения роста цепи зависит от температуры. Обрыв цепи происходит в результате тех же реакций, что и при анионной полимеризации, в частности реакции переноса гидрид-иона на мономер или противоион. Кроме того молекулярную массу образующегося полимера можно регулировать также введением в реакционную среду агентов передачи цепи - водорода и алюминийгидрида.

Слайд 143
Скорость роста цепи может быть выражена кинетическим уравнением, похожим на подобное
где [I] и [M] - концентрации инициатора и мономера, а α и β -порядки реакции по инициатору и мономеру.
Катализаторы Циглера-Натта широко используются для полимеризации этилена, пропилена, диенов и некоторых полярных и гетероциклических мономеров.
В промышленности методом анионно-координационной полимеризации получают стереорегулярные каучуки (СКИ-3, СКИ-5, СКД) и полиолефины (полиэтилен низкой плотности - ПЭНП) и др.

Слайд 144
ПОЛИКОНДЕНСАЦИЯ
Поликонденсация - это синтез полимеров взаимодействием би- или полифункциональных мономеров или
Реакционными (активными) центрами при поликонденсации можно считать функциональные группы.
Процессы поликонденсации играют большую роль в природе и технике. Поликонденсация лежит в основе образования всех природных ВМС: белков, целлюлозы, крахмала, нуклеиновых кислот и др.
Первое промышленное производство синтетического полимера - феноло-формальдегидной смолы (Лео Бакеланд, 1909), основано на реакции поликонденсации.

Слайд 145
Большой вклад в развитие знаний о процессах поликонденсации внесли отечественнве ученые:

Слайд 146
КЛАССИФИКАЦИЯ И ТЕРМИНОЛОГИИ
Поликонденсация бифункциональных мономеров называется линейной.
Поликонденсация, в которой участвует
В каждом случае образуются, соответственно, линейные и сетчатые полимеры
Важная разновидность поликонденсации - полициклоконденсация, при которой продукт линейной поликонденсации подвергается внутримолекулярной циклизации (протекает в две стадии: линейная поликонденсация, а затем собственно циклизация). Применяется для получения термостойких полимеров.

Слайд 147
По типу (и числу) участвующих в реакции мономеров различают:
- гомополиконденсацию (участвует
- гетерополиконденсацию, когда взаимодействуют мономеры с различными функциональными группами, например, получение сложного полиэфира из гликолей и двухосновных кислот (иногда встречается термин - сополиконденсация).
Поликонденсация - ступенчатый процесс, при котором мономеры, взаимодействуя друг с другом, исчерпываются на сравнительно ранней стадии реакции, а высокомолекулярный полимер образуется обычно в результате реакции ранее образовавшихся олигомеров и полимерных цепей при глубине превращения функциональных групп, близкой к 100% - в отличии от полимеризации, там высокомолекулярные продукты образуются при невысоких степенях конверсии мономера. Причем промежуточные олигомерные продукты при поликонденсации можно выделить
 участвующих в реакции мономеров различают:- гомополиконденсацию (участвует один мономер с различными")
Слайд 148
При поликонденсации часто возможны обратная и различные обменные реакции. В зависимости
К первой условно относят поликонденсацию с константой равновесия (Кр ) больше 103, ко второй - с Кр менее 103 (обычно 0.1-10). При неравновесной поликонденсации обычно образуются значительно более высокомолекулярные полимеры (ММ > 100 000), чем при равновесной (ММ ~ 20 000-30 000).
МОНОМЕРЫ
В качестве мономеров в поликонденсации используют соединения, содержащие в молекуле не менее двух функциональных групп. Их можно разделить на три основных типа:

Слайд 149
1) Мономеры (например, диамины или дихлорангидриды дикарбоновых кислот), содержащие в молекулах
2) Мономеры (например, гидроксикислоты или аминокислоты), содержащие различные функциональные группы, которые способны реагировать друг с другом, приводя к образованию полимера, например:
3) Мономеры, содержащие одинаковые функциональные группы, способные реагировать между собой в данных условиях, например, гликоли, поликонденсация которых приводит к образованию простых полиэфиров:
 Мономеры (например, диамины или дихлорангидриды дикарбоновых кислот), содержащие в молекулах одинаковые функциональные группы, не")
Слайд 150
В условиях поликонденсации возможны случаи, когда функциональные группы одного или нескольких
Функциональность и активность функциональных групп мономера зависит от его строения, условий поликонденсации и природы реагирующего с ним мономера. При этом из определенных полифункциональных мономеров (мономеры с числом функциональных групп больше двух) получают как линейные, так и сетчатые полимеры.

Слайд 151Примаер: при поликонденсации тетрафункциональных мономеров -бис-(о-фенилендиаминов) с бис-(α-дикетонами) образуются линейные, плавкие
тогда как при реакции этих же тетрафункциональных мономеров с дихлорангидридами дикарбоновых кислот получаются разветвленные и сетчатые полиамиды
Поэтому различают возможную, практическую и относительную (определяется как отношение практической функциональности к возможной) функциональности мономеров.
 с бис-(α-дикетонами) образуются линейные, плавкие и растворимые полифенилхиноксалинытогда как")
Слайд 152
Часто ограничиваются рассмотрением средней функциональности:
т.е. среднее количество функциональных групп, приходящихся на
Для поликонденсации чаще всего используются мономеры с амино-, кар-бокси-, гидрокси-, меркаптогруппами, из которых получают важнейшие типы поликонденсационных полимеров.
В отличие от полимеризации, при которой для получения определенного полимера обычно требуется один мономер, поликонденсационные полимеры одного типа можно синтезировать из мономеров с самыми разнообразными функциональными группами. В принципе можно использовать любые реакции образования химических связей С-С, С-О и др.

Слайд 153
Например, при получении сложных полиэфиров гидроксилсодержащие мономеры могут быть заменены на
Естественно, что при такой замене меняются закономерности и условия поликонденсации, тип катализа, характер концевых групп в образующихся макромолекулах и, кроме того, появляются возможности получения полимеров с заданным комплексом технологических и эксплуатационных свойств.
ОСНОВНАЯ И ПОБОЧНЫЕ РЕАКЦИИ
В общем виде схема основной реакции поликонденсации - роста цепи - может быть представлена следующим образом:
где n и m - любое целое число, включая единицу, Х и Y-исходные функциональные группы, xy - низкомолекулярный продукт поликонденсации.

Слайд 154
При этом взаимодействие мономеров или образовавшихся олигомеров друг с другом (и
Поскольку в отличие от полимеризации, при поликонденсации мономеры исчерпываются уже при невысоких степенях завершенности реакции, рост цепи высокомолекулярного полимера происходит преимущественно в результате многократного соединения между собой олигомерных или полимерных молекул по концевым функциональным группам (принцип многократного удвоения), при этом число молекул в системе уменьшается (в этом ступенчатый характер поликонденсации).
Уменьшается в ходе поликонденсации и количество исходных функциональных групп - реакционных (активных) центров, хотя в ряде случаев образующиеся при поликонденсации связи могут разрываться под действием исходных реакционных центров или под действием функциональых групп низкомолекулярных продуктов поликонденации.

Слайд 155
Таким образом росту полимерной цепи при равновесной поликонденсации сопутствует обратная реакция
При поликонденсации функциональные группы мономеров, олигомеров и полимерных цепей расходуются не только на рост цепи, но и на побочные реакции (реакции с монофункциональными примесями или со специально введенными в процесс веществами, могут менять свою функциональность группы, например, декарбоксилирование карбоновых кислот и др.), что также лимитирует молекулярную массу образующегося полимера.
При поликонденсации возможны также циклизация и обменные реакции.
Циклизация может быть внутримолекулярной, когда кольца образуются при реакции функциональных групп одной молекулы, или межмолекулярной при взаимодействии двух или более молекул одинаковой или различной природы, например:

Слайд 156
Возможность циклизации определяется соотношением двух факторов:
1) снижения вероятности образования цикла по
2) напряженности цикла, которая уменьшается при увеличении его размера вплоть до 6-членного, затем возрастает при изменении числа членов в цикле от 6 до 9-11, а затем вновь снижается при переходе к еще большим циклам.
 снижения вероятности образования цикла по мере увеличения его размера")
Слайд 157
В результате совместного действия обоих этих факторов при получении методом поликонденсации
Обменные реакции особенно эффективны при повышенных температурах поликонденсации. Их делят на два основных типа:
1) реакции обмена образовавшихся при поликонденсации групп (сложноэфирной, амидной или др.) и даже некоторых циклов (например, имидного) с функциональными группами мономеров или примесей (например, алкоголиз, ацидолиз, аминолиз);
2) реакции межцепного обмена между образовавшимися при полимеризации одно- или разнотипными группами (например, эфиролиз, амидолиз).
Эффективность обменных реакций зависит от соотношения скоростей основной и побочных реакций. Обменные реакции могут существенно влиять на молекулярную массу и ММР поликонденсационного полимера, микроструктуру сополимера.

Слайд 158
В ряде случаев обменные реакции положены в основу получения поликонденсационных гомо-
Ограничение роста полимерной цепи может быть обусловлено и чисто физическими причинами, например, преждевременным выпадением полимера из реакционной среды в осадок при поликонденсации в растворе (особенно если это сопровождается его кристаллизацией), однако если выпадающий из раствора полимер набухает в реакционной среде, рост цепи часто не прекращается.

Слайд 159
КИНЕТИКА, КАТАЛИЗ, МОЛЕКУЛЯРНО-МАССОВОЕ РАСПРЕДЕЛЕНИЕ
Своеобразие поликонденсации, существенно отличающее ее от полимеризации, заключается
исчезновение мономеров в реакционной среде, как уже отмечалось, наступает задолго до образования полимера достаточно высокой степени полимеризации, или молекулярной массы;
в большинстве случаев (при поликонденсации в гомогенных условиях) получение полимера высокой молекулярной массы возможно лишь при очень высокой (близкой к количественной) степени завершенности реакции (глубине превращения).
Это наглядно иллюстрирует следующая зависимость средней степени полимеризации (
) полимера от степени завершенности реакции (х):

Слайд 160
Поэтому особенно важно изучение кинетики поликонденсации при протекании в гомо- или
Кинетика поликонденсации, включающей бесконечное число актов роста цепи, как правило, с трудом поддается количественному анализу. Для его упрощения вводят следующие допущения (допущения Флори):
1) реакционные способности обеих однотипных функциональных групп бифункционального мономера одинаковы;
2) реакционная способность одной функциональной группы бифункционального мономера не зависит от того, прореагировала или нет другая группа;
3) реакционная способность функциональной группы не зависит от размера молекулы, с которой она связана.
При этом описание кинетики поликонденсации становится таким же, как кинетики аналогичных реакций низкомолекулярных соединений.

Слайд 161
Во многих случаях указанные допущения перестают быть справедливыми: имеются бифункциональные мономеры,
Кинетику поликонденсации описывают обычно до степени завершенности реакции 0,90-0,95, т.е. когда высокомолекулярный полимер в реакционной системе практически еще отсутствует.
Кинетическое исследование реакций поликонденсации, в которых допущения Флори не выполняются, а также заключительных стадий поликонденсации (собственно реакций полимерообразования), когда существенную роль начинают играть диффузионные факторы, требует применения более сложных кинетических уравнений и систем уравнений, которые численно решаются с помощью вычислительной техники.

Слайд 162
Кинетическая и другие характеристики равновесной и неравновесной поликонденсации сильно различаются.
Равновесная
Неравновесная поликонденсация обычно протекает с высокой скоростью [константа скорости до 105 л/(моль⋅с)], как правило, она сильно экзотермична и характеризуется низкими значениями энергии активации (8-40 кДж/моль).
Максимально возможная молекулярная масса полимера, получаемого равновесной поликонденсацией определяется термодинамическими факторами и может быть определена по уравнению:
где [xy] - концентрация низкомолекулярного продукта поликонденсации в реакционной системе.

Слайд 163
Из этого соотношения следует, что для получения высокомолекулярных полимеров поликонденсацию следует
В неравновесной поликонденсации молекулярная масса полимера не определяется термодинамическими факторами; большое значение для получения высокомолекулярных полимеров в этом процессе имеет уменьшение вклада побочных реакций.
Например, как влияют условия синтеза полиамида на его молекулярную массу:

Слайд 164
Процессы поликонденсации можно проводить как в одну, так и в несколько
Кинетика поликонденсации в гетерогенных системах, когда появляется граница раздела, должна учитывать распределение между фазами реагентов, их поверхностную активность, различные процессы массопереноса и другие факторы. Таким образом, кинетика поликонденсации (межфазная поликонденсация) может определяться не только химическим взаимодействием функциональных групп, но и диффузионными факторами.
Для ускорения поликонденсации используют различные приемы:
- активацию функциональных групп (например, замена карбоксильных групп на хлорангидридные или сложноэфирные группы, содержащие остатки сильно кислых фенолов, например, нитрофенола);
- применение активных растворителей (ДМФА, ДМСО, N,N-диметил-ацетамида, N-метилпирролидона);

Слайд 165
- введение активирующих агентов (например, пиридина и трифенилфосфита при поликонденсации дикарбоновых
- катализ.
Катализаторами служат карбоновые кислоты, их соли, алкоголяты, третичные амины, сложные эфиры, фосфорные кислоты и многие другие соединения или их смеси, ускоряющие отдельные стадии поликонденсации, например, синтез полиэтилентерефталата в присутствии ацетатов металлов и Sb2O3.
Некоторые соединения, например краун-эфиры, четвертичные аммониевые и фосфониевые соли, применяемые для катализа поликонденсации, протекающей в гетерогенных условиях, являются катализаторами межфазного переноса, облегчая перенос реагентов из одной фазы в другую, например, из водной в органическую.
Процессы поликонденсации могут быть:
автокаталитическими (синтез полиамидокислот из диангидридов тетракарбоновых кислот и диаминов, катализируемый полиамидокислотой),
- самокатализируемыми (синтез сложных полиэфиров из дикарбоновых кислот и диолов, катализируемый дикарбоновыми кислотами.
;- катализ.Катализаторами")
Слайд 166
Катализаторы применяют как в истинно каталитических количествах (молярные проценты), так и
ММР полимера, получаемого линейной поликонденсацией, часто описывается уравнением:
где Pn - молярная или численная доля х-меров.
Это распределение обычно называется наиболее вероятным или распределением Флори.
Необходимым условием образования высокомолекулярного полимера при линейной поликонденсации является стехиометрическое соотношение реагирующих функциональных групп.
, так и в значительно больших (например,")
Слайд 167
В этом случае справедливо уравнение Карозерса:
Например, при поликонденсации глицерина и фталевой
а
при
т.е. при этой глубине превращения возможно образование сшитого полимера!
:")
Слайд 168
Таким образом
Если
, т.е. полимер не образуется, а только димер.
При
, т.е. при х=0.9
, а при х=0.998
.
При отклонении от стехиометрического соотношения обычно образуются более низкомолярные полимеры. Степень полимеризации полимера, синтезируемого при избытке (или недостатке) одного из мономеров, может быть определена по уравнению:
где r -стехиометрический разбаланс, т.е. соотношение количеств функциональных групп обоих мономеров.

Слайд 169
При х = 1 указанное уравнение принимает вид:
Поликонденсация при заданном избытке
Так поступают, например, при синтезе полиуретанов; вначале из дикарбоновых кислот и избытка гликолей получают сложные полиэфиры с концевыми группами ОН, а затем проводят их взаимодействие с диизоцианатами.
Примеси монофункциональных соединений также уменьшают степень полимеризации:
где N - мольная доля монофункциональных соединений по отношению к бифункциональным соединениям одинаковой химической природы.













