№6
по курсу «Анализ и контроль
качества лекарственных средств»
- Главная
- Разное
- Дизайн
- Бизнес и предпринимательство
- Аналитика
- Образование
- Развлечения
- Красота и здоровье
- Финансы
- Государство
- Путешествия
- Спорт
- Недвижимость
- Армия
- Графика
- Культурология
- Еда и кулинария
- Лингвистика
- Английский язык
- Астрономия
- Алгебра
- Биология
- География
- Детские презентации
- Информатика
- История
- Литература
- Маркетинг
- Математика
- Медицина
- Менеджмент
- Музыка
- МХК
- Немецкий язык
- ОБЖ
- Обществознание
- Окружающий мир
- Педагогика
- Русский язык
- Технология
- Физика
- Философия
- Химия
- Шаблоны, картинки для презентаций
- Экология
- Экономика
- Юриспруденция
Хроматографические методы анализа и их применение для контроля качества лекарственных средств (продолжение) презентация
Содержание
- 1. Хроматографические методы анализа и их применение для контроля качества лекарственных средств (продолжение)
- 2. Жидкостная хроматография метод разделения, в котором подвижная
- 4. Оборудование (принципиальная схема)
- 5. Варианты проведения ЖХ 1. Изократический режим –
- 6. Хроматографические колонки Стальные трубки внутренним диаметром
- 7. Неподвижные фазы Общие требования: 1. Для обеспечения
- 8. Неподвижные фазы Классификация: 1. Полярные фазы (для
- 9. Выбор подвижной фазы для нормально-фазовой хроматографии
- 10. Неподвижные фазы 2. Среднеполярные фазы (для обращенно-фазовой
- 11. Неподвижные фазы 3. Неполярные фазы (для обращенно-фазовой
- 13. Принципы разделения 1. Полярные фазы –
- 14. Принципы разделения 2. Средне- и неполярные фазы:
- 15. Механизмы удерживания
- 16. Закономерности удерживания в обращенно-фазовой ВЭЖХ Вытеснительная модель
- 17. Влияние рН ПФ и температуры На неполярных
- 18. Детекторы в ЖХ 1. Детектор спектрофотометрический (область
- 19. 2. Детектор на основе диодной матрицы –
- 20. СФ-детектор Детектор на основе диодной матрицы
- 21. Флуориметрический детектор Селективный детектор, основанный на измерении
- 22. Рефрактометрический детектор Основан на измерении величины преломления света элюата (универсальный детектор)
- 23. Электрометрические детекторы 1. Амперометрический детектор (детекция органических
- 24. Масс-спектрометрическое детектирование Универсальный (полный ионный ток) и селективный (сканирование индивидуальных масс ионов) детектор
- 26. Масс-спектрометрическое детектирование 1. Ионизация образца (ПФ + разделенные вещества) Принципы: 1.1. электрораспылительная (ESI)
- 28. Примеры ионизации
- 29. Масс-спектрометрическое детектирование 1.2. Химическая ионизация при атмосферном давлении:
- 30. 1.3.MALDI (ионизация лазерной десорбцией при взаимодействии с матрицей)
- 31. MALDI-TOF (ионизация лазерной десорбцией при взаимодействии с матрицей и времяпролетным детектированием)
- 32. Масс-анализаторы А. непрерывные масс-анализаторы 1. Магнитный и
- 33. Масс-спектрометрическое детектирование Достоинства: 1. Высочайшая чувствительность органических
- 34. Сравнение различных типов детекторов
- 35. Применение ВЭЖХ в фармацевтическом анализе 1. Идентификация
- 36. Применение ВЭЖХ в фармацевтическом анализе 1.2. Идентификация по времена удерживания и спектрам поглощения пиков.
- 37. Применение ВЭЖХ в фармацевтическом анализе 1.3. Идентификация
- 38. Применение ВЭЖХ в фармацевтическом анализе 2. Определение
- 39. 2. Определение специфических (родственных) примесей – полуколичественный
- 40. 2. Определение специфических (родственных) примесей – полуколичественный анализ 2.3. Методом внутренней нормализации
- 41. Определение специфических (родственных) примесей 2.4. Количественное определение (например, токсичные примеси) – методом градуировочного графика
- 42. 2.5. Определение энантиомерной чистоты
- 43. Применение ВЭЖХ в фармацевтическом анализе 3. Определение
- 44. Капиллярный электрофорез Метод капиллярного электрофореза (КЭ) основан
- 45. Капиллярный электрофорез МЭКХ - вариант капиллярного электрофореза,
- 46. После подачи к концам капилляра высокого напряжения
- 47. Основные параметры КЭ 1. Время миграции (tм)
- 49. Механизм ЭОП
- 50. Электроосмотический поток Уникальной особенностью ЭОП является плоский
- 51. Капилляры для разделения Подавляющее большинство разделений в
- 52. Ввод образца Проба может быть введена в
- 53. Детектирование
- 54. Пример разделения неорганических ионов (КЭ) 1 –
- 56. Термические методы анализа Основаны на установлении зависимостей
- 69. БЛАГОДАРЮ ЗА ВНИМАНИЕ!
Слайд 1Хроматографические методы анализа и их применение для контроля качества лекарственных средств
(продолжение)
Лекция
Лекция №6 по курсу")
Слайд 2Жидкостная хроматография
метод разделения, в котором подвижная фаза представляет собой жидкость, а
неподвижная – твердую или жидкую фазу (не смешивающуюся с подвижной фазой).
Различают:
А. колоночную (низкого и высокого давления – ВЭЖХ или ЖХВД) и планарную (ТСХ) хроматографии.
Б. по полярности неподвижной/подвижной фаз и по механизму удерживания/разделения:
- прямо-фазовую ЖХ (НФ – полярная, ПФ – неполярные жидкости)
- обращенно-фазовую ЖХ (НФ – неполярная или среднеполярная, ПФ – полярные жидкости).
- ионообменная или ионная.
- эксклюзионная (гель-хроматография).
Различают:
А. колоночную (низкого и высокого давления – ВЭЖХ или ЖХВД) и планарную (ТСХ) хроматографии.
Б. по полярности неподвижной/подвижной фаз и по механизму удерживания/разделения:
- прямо-фазовую ЖХ (НФ – полярная, ПФ – неполярные жидкости)
- обращенно-фазовую ЖХ (НФ – неполярная или среднеполярная, ПФ – полярные жидкости).
- ионообменная или ионная.
- эксклюзионная (гель-хроматография).


")
Слайд 5Варианты проведения ЖХ
1. Изократический режим – постоянный состав ПФ в течение
всего анализа (разделение родственных веществ, мало отличающихся по полярности).
2. Градиентный режим – изменяется состав ПФ (линейный градиент – с постоянной скоростью и нелинейный градиент – изменение состава с переменной скоростью) – для разделения веществ, отличающихся по полярности (например, смесей водо- и жирорастворимых витаминов), для повышения эффективности разделения.
2. Градиентный режим – изменяется состав ПФ (линейный градиент – с постоянной скоростью и нелинейный градиент – изменение состава с переменной скоростью) – для разделения веществ, отличающихся по полярности (например, смесей водо- и жирорастворимых витаминов), для повышения эффективности разделения.

Слайд 6Хроматографические колонки
Стальные трубки внутренним диаметром 2-5 мм, длиной 5-30 см,
с пористыми фильтрами с обоих концов. Для защиты аналитической колонки используются сменные предколонки (длиной 1-2 см).

Слайд 7Неподвижные фазы
Общие требования:
1. Для обеспечения высокой эффективности разделения – размер частиц
сорбента должен быть четко установленного размера (1,8 – 10 мкм), четко сферической формы.
2. Должен быть устойчивым к повышенному давлению (нехрупким), к химическим веществам (устойчивость при рН 2-8) и температуре (до 60-80оС).
3. Обладать высокой удельной поверхностью (60-300 м2/г) и определенным размером пор (10-300 нм).
4. Обратимая сорбция разделенных соединений.
2. Должен быть устойчивым к повышенному давлению (нехрупким), к химическим веществам (устойчивость при рН 2-8) и температуре (до 60-80оС).
3. Обладать высокой удельной поверхностью (60-300 м2/г) и определенным размером пор (10-300 нм).
4. Обратимая сорбция разделенных соединений.

Слайд 8Неподвижные фазы
Классификация:
1. Полярные фазы (для прямофазной ЖХ) – немодифицированные силикагели, аминопропилсилилсиликагели,
диольные производные силилсиликагеля, иониты (HILIC)
 – немодифицированные силикагели, аминопропилсилилсиликагели, диольные производные силилсиликагеля, иониты (HILIC)")

Слайд 10Неподвижные фазы
2. Среднеполярные фазы (для обращенно-фазовой ЖХ) – химически модифицированные силикагели
с привитыми цианопропильными (СN), пентафторбензильными (PFP), фенильными (Ph), фенил-гексильными группами.
 – химически модифицированные силикагели с привитыми цианопропильными (СN),")
Слайд 11Неподвижные фазы
3. Неполярные фазы (для обращенно-фазовой ЖХ) – химически модифицированные силикагели
с привитыми октильными (С8), октадецильными (С18), фенильными и октадецильными (С18-Ph), и др.
 – химически модифицированные силикагели с привитыми октильными (С8),")

Слайд 13Принципы разделения
1. Полярные фазы –
1.1. для разделения неполярных и малополярных
веществ (слабо адсорбируются НФ) – малополярные элюенты (гексан (гептан) + низкая доля полярного растворителя, дихлорметан + низкая доля полярного растворителя).
1.2. для разделения полярных веществ (сильно адсорбируются НФ) – высокая доля полярных растворителей (метанол, ацетонитрил, вода), необходимо устанавливать рН (от 2 до 9), добавлять буферный раствор (ионная сила).
1.2. для разделения полярных веществ (сильно адсорбируются НФ) – высокая доля полярных растворителей (метанол, ацетонитрил, вода), необходимо устанавливать рН (от 2 до 9), добавлять буферный раствор (ионная сила).
")
Слайд 14Принципы разделения
2. Средне- и неполярные фазы:
2.1. Для разделения неполярных веществ используются
ПФ с высоким (40-100%) содержанием органического растворителя (ацетонитрил, метанол, тетрагидрофуран).
2.2. Для разделения ионизируемых органических веществ (кислот, оснований, амфолитов, ионов) необходимо использовать буферные растворы (рН 2-8 или для ряда колонок – 1-10).
2.3. Для разделения органических ионов и сильнополярных веществ – прибавляют ион-парные реагенты (алкилсульфонаты, четвертичные аммониевые соли).
2.2. Для разделения ионизируемых органических веществ (кислот, оснований, амфолитов, ионов) необходимо использовать буферные растворы (рН 2-8 или для ряда колонок – 1-10).
2.3. Для разделения органических ионов и сильнополярных веществ – прибавляют ион-парные реагенты (алкилсульфонаты, четвертичные аммониевые соли).
")

Слайд 16Закономерности удерживания в обращенно-фазовой ВЭЖХ
Вытеснительная модель удерживания:
1. Из ПФ на
поверхности неполярного/среднеполярного сорбента адсорбируется органический компонент (метанол, ацетонитрил).
2. При введении органического вещества оно вытесняет с поверхности сорбента часть молекул органического модификатора. Данный процесс обратимый и при движении новой порции ПФ органический модификатор вновь вытесняет сорбат.
2. При введении органического вещества оно вытесняет с поверхности сорбента часть молекул органического модификатора. Данный процесс обратимый и при движении новой порции ПФ органический модификатор вновь вытесняет сорбат.

Слайд 17Влияние рН ПФ и температуры
На неполярных фазах за счет дисперсионных взаимодействий
лучше удерживаются неионизированные молекулы. При ионизации молекул удерживание при прочих равных условиях уменьшается.
Температура незначительно влияет на удерживание органических молекул в водно-органических фазах. При повышении температуры уменьшается вязкость ПФ и давление на колонке (возможно работать на более высоких скоростях ПФ).
Температура незначительно влияет на удерживание органических молекул в водно-органических фазах. При повышении температуры уменьшается вязкость ПФ и давление на колонке (возможно работать на более высоких скоростях ПФ).

Слайд 18Детекторы в ЖХ
1. Детектор спектрофотометрический (область длин волн – УФ 190-360
нм (дейтериевая лампа), видимая – 360-900 нм – галогеновая лампа) – работает при фиксированных длинах волн (от 1 до 10 и более) – двумерная хроматография.
, видимая")
Слайд 192. Детектор на основе диодной матрицы – сканирование оптической плотности элюата
в заданном диапазоне длин волн с большой скоростью (трехмерная хроматограмма – время-длина волны-сигнал детектора – е.о.п.).


Слайд 21Флуориметрический детектор
Селективный детектор, основанный на измерении интенсивности флуоресценции разделенных веществ (устанавливаются
длины волн возбуждения и эмиссии (испускания)).

Слайд 22Рефрактометрический детектор
Основан на измерении величины преломления света элюата (универсальный детектор)
")
Слайд 23Электрометрические детекторы
1. Амперометрический детектор (детекция органических веществ, обладающих ОВ-свойствами) – может
быть комплексный с ферментативными реакциями.
2. Кондуктометический детектор (детекция ионов, основанная на измерении проводимости подвижной фазы).
3. Кулонометрический детектор.
2. Кондуктометический детектор (детекция ионов, основанная на измерении проводимости подвижной фазы).
3. Кулонометрический детектор.
 – может быть комплексный с ферментативными")
Слайд 24Масс-спектрометрическое детектирование
Универсальный (полный ионный ток) и селективный (сканирование индивидуальных масс ионов)
детектор
 и селективный (сканирование индивидуальных масс ионов) детектор")

Слайд 26Масс-спектрометрическое детектирование
1. Ионизация образца (ПФ + разделенные вещества)
Принципы:
1.1. электрораспылительная (ESI)
Принципы:1.1. электрораспылительная (ESI)")



")
Слайд 31MALDI-TOF (ионизация лазерной десорбцией при взаимодействии с матрицей и времяпролетным детектированием)
")
Слайд 32Масс-анализаторы
А. непрерывные масс-анализаторы
1. Магнитный и электростатический секторный масс-анализатор (Sector)
2. Квадрупольный масс-анализатор (Quadrupole mass
analyzer)
Б. импульсные масс-анализаторы
1. Времяпролётный масс-анализатор (Time-of-flight mass spectrometry)
2. Ионная ловушка (Ion trap)
3. Квадрупольная линейная ловушка (Quadrupole ion trap)
4. Масс-анализатор ионно-циклотронного резонанса с Фурье-преобразованием (Fourier transform ion cyclotron resonance)
5. Орбитрэп (Orbitrap)
Б. импульсные масс-анализаторы
1. Времяпролётный масс-анализатор (Time-of-flight mass spectrometry)
2. Ионная ловушка (Ion trap)
3. Квадрупольная линейная ловушка (Quadrupole ion trap)
4. Масс-анализатор ионно-циклотронного резонанса с Фурье-преобразованием (Fourier transform ion cyclotron resonance)
5. Орбитрэп (Orbitrap)
2. Квадрупольный масс-анализатор (Quadrupole mass analyzer)Б. импульсные масс-анализаторы1. Времяпролётный")
Слайд 33Масс-спектрометрическое детектирование
Достоинства:
1. Высочайшая чувствительность органических веществ, биополимеров (10-15 г/пробе).
2. Высокая специфичность
детекции (последовательная масс-спектрометрия (дочерних ионов)).
3. Метод сбора информации о структуре молекул (точность установления молярных масс – до 0,0001-0,000001 а.е.м.).
4. Широкий линейный диапазон – 106-107.
5. Наличие баз данных по масс-спектрам огромного числа органических веществ (для ГХ/МС).
6. Основной детектор при проведении биоэквивалентных испытаний, допинг-контроля, судебно-химической экспертизы, исследования метаболизма, генеза БАВ и др.
Недостатки:
1. Сложность и высокая стоимость оборудования и расходных материалов.
2. Необходимо дополнительное обучение непосредственно масс-спектрометрии и интерпретации спектров.
3. Метод сбора информации о структуре молекул (точность установления молярных масс – до 0,0001-0,000001 а.е.м.).
4. Широкий линейный диапазон – 106-107.
5. Наличие баз данных по масс-спектрам огромного числа органических веществ (для ГХ/МС).
6. Основной детектор при проведении биоэквивалентных испытаний, допинг-контроля, судебно-химической экспертизы, исследования метаболизма, генеза БАВ и др.
Недостатки:
1. Сложность и высокая стоимость оборудования и расходных материалов.
2. Необходимо дополнительное обучение непосредственно масс-спектрометрии и интерпретации спектров.
.2. Высокая специфичность детекции (последовательная масс-спектрометрия (дочерних")

Слайд 35Применение ВЭЖХ в фармацевтическом анализе
1. Идентификация веществ
1.1. Сравнение времен удерживания со
стандартным веществом
Раствор сравнения
Испытуемый раствор

Слайд 36Применение ВЭЖХ в фармацевтическом анализе
1.2. Идентификация по времена удерживания и спектрам
поглощения пиков.

Слайд 37Применение ВЭЖХ в фармацевтическом анализе
1.3. Идентификация веществ (образцов) по хроматографическому профилю
(наличие определенного числа пиков с относительными временами удерживания по любому из компонентов) – растительные экстракты, ЛС сложного состава.
 по хроматографическому профилю (наличие определенного числа пиков")
Слайд 38Применение ВЭЖХ в фармацевтическом анализе
2. Определение специфических (родственных) примесей – полуколичественный
анализ
2.1. По стандартному образцу примеси
2.1. По стандартному образцу примеси
 примесей – полуколичественный анализ2.1. По стандартному образцу примеси")
Слайд 392. Определение специфических (родственных) примесей – полуколичественный анализ
2.2. По стандартному раствору
основного вещества, разведенному до определенного предела (0,05-5%).
 примесей – полуколичественный анализ2.2. По стандартному раствору основного вещества, разведенному до")
Слайд 402. Определение специфических (родственных) примесей – полуколичественный анализ
2.3. Методом внутренней нормализации
 примесей – полуколичественный анализ2.3. Методом внутренней нормализации")
Слайд 41Определение специфических (родственных) примесей
2.4. Количественное определение (например, токсичные примеси) – методом
градуировочного графика
 примесей2.4. Количественное определение (например, токсичные примеси) – методом градуировочного графика")

Слайд 43Применение ВЭЖХ в фармацевтическом анализе
3. Определение основных показателей готовых лекарственных средств
– однородность дозированных единиц, тест «Растворение», количественное определение стабилизаторов, консервантов, красителей и др.
4. Определение пластификаторов в упаковочных материалах.
5. Определение остаточных количеств пестицидов (гербицидов, инсектицидов) в ЛРС, продуктах из ЛРС.
6. Определение остаточных количеств активных фармацевтических ингредиентов на оборудовании (контроль отмывки оборудования), в сточных водах.
4. Определение пластификаторов в упаковочных материалах.
5. Определение остаточных количеств пестицидов (гербицидов, инсектицидов) в ЛРС, продуктах из ЛРС.
6. Определение остаточных количеств активных фармацевтических ингредиентов на оборудовании (контроль отмывки оборудования), в сточных водах.

Слайд 44Капиллярный электрофорез
Метод капиллярного электрофореза (КЭ) основан на разделении заряженных компонентов сложной
смеси в кварцевом капилляре под действием приложенного электрического поля за счёт подачи высокого напряжения к концам капилляра.
Наиболее распространёнными вариантами метода КЭ являются: 1 капиллярный зонный электрофорез (КЗЭ)
2. мицеллярная электрокинетическая хроматография (МЭКХ).
КЗЭ - метод разделения, реализуемый в капиллярах и основанный на различии в электрокинетических подвижностях заряженных частиц как в водных, так и в неводных электролитах.
Наиболее распространёнными вариантами метода КЭ являются: 1 капиллярный зонный электрофорез (КЗЭ)
2. мицеллярная электрокинетическая хроматография (МЭКХ).
КЗЭ - метод разделения, реализуемый в капиллярах и основанный на различии в электрокинетических подвижностях заряженных частиц как в водных, так и в неводных электролитах.
 основан на разделении заряженных компонентов сложной смеси в кварцевом капилляре")
Слайд 45Капиллярный электрофорез
МЭКХ - вариант капиллярного электрофореза, который позволяет проводить разделение соединений
ионного и нейтрального характера при использовании ПАВ. Разделение электро-нейтральных соединений осуществляется благодаря введению в состав ведущего электролита поверхностно-активных веществ - мицеллообразователей. Чаще всего используют анионный ПАВ (например, ДДС) в концентрациях, превышающих критическую концентрацию мицелообразования, что приводит к формированию так называемой «псевдостационарной фазы», и аналиты распределяются между мицеллой и буферным электролитом согласно их гидрофобности.

Слайд 46После подачи к концам капилляра высокого напряжения (до 30 кВ), компоненты
смеси начинают двигаться по капилляру с разной скоростью, зависящей в первую очередь от заряда и величины ионного радиуса и, соответственно, в разное время достигают зоны детектирования. Полученная последовательность пиков называется электрофореграммой, при этом качественной характеристикой вещества является параметр удерживания (время миграции), а количественной – высота или площадь пика, пропорциональная концентрации вещества.
, компоненты смеси начинают двигаться по")
Слайд 47Основные параметры КЭ
1. Время миграции (tм) - время, необходимое компоненту для
прохождения им эффективной длины капилляра (Lэфф) от зоны ввода пробы (начала капилляра) до зоны детектирования;
2. Электроосмотический поток (ЭОП) - течение жидкости в капилляре под действием приложенного электрического поля. Время, необходимое жидкости для преодоления эффективной длины капилляра вследствие возникающего ЭОП, называют временем ЭОП (tэоп) и экспериментально определяют из электрофореграммы по времени миграции нейтрального компонента – маркера ЭОП.
3. Подвижность ЭОП (μэоп) - представляет собой отношение скорости ЭОП к напряженности электрического поля. Скорость ЭОП положительна при направлении движения жидкости от входного участка капилляра к детектору и отрицательна при обратном направлении. В свою очередь, скорость ЭОП вычисляют по формуле:
νэоп= Lэфф / tэоп.
2. Электроосмотический поток (ЭОП) - течение жидкости в капилляре под действием приложенного электрического поля. Время, необходимое жидкости для преодоления эффективной длины капилляра вследствие возникающего ЭОП, называют временем ЭОП (tэоп) и экспериментально определяют из электрофореграммы по времени миграции нейтрального компонента – маркера ЭОП.
3. Подвижность ЭОП (μэоп) - представляет собой отношение скорости ЭОП к напряженности электрического поля. Скорость ЭОП положительна при направлении движения жидкости от входного участка капилляра к детектору и отрицательна при обратном направлении. В свою очередь, скорость ЭОП вычисляют по формуле:
νэоп= Lэфф / tэоп.
 - время, необходимое компоненту для прохождения им эффективной длины")
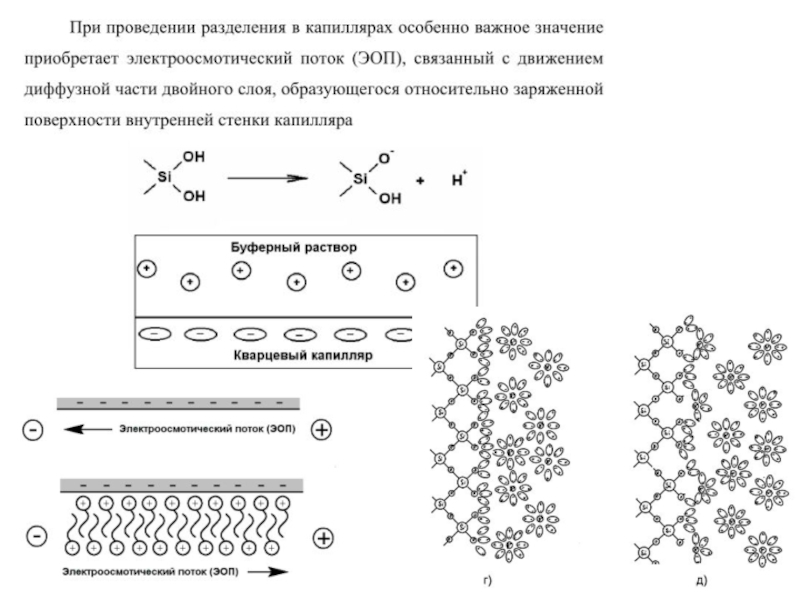

Слайд 50Электроосмотический поток
Уникальной особенностью ЭОП является плоский профиль потока в капилляре. Такой
профиль выгоден, поскольку уменьшается размывание зон разделяемых веществ. Следует отметить, что эффективность разделения в капиллярном электрофорезе прямо пропорциональна, а время анализа – обратно пропорционально напряжению, приложенному к электродам. Разделение в КЭ может быть выполнено как с положительной, так и отрицательной полярностью электродов. Зная значения рКа для компонентов пробы, можно выбрать буфер с подходящим значением рН и полярность электродов, чтобы образец двигался в сторону детектора. Скорость миграции зависит от напряженности электрического поля, которая обычно составляет 200-400 В/см.

Слайд 51Капилляры для разделения
Подавляющее большинство разделений в КЭ проводят с использованием кварцевых
капилляров имеющих внешнее полимерное покрытие, обычно - полиимидное, улучшающее их механическую прочность, и значительно реже полимерные капилляры, например из тефлона. Внутренний диаметр капилляров колеблется в пределах от 25 до 200 микрон, а длина капилляра в зависимости от поставленной задачи – от нескольких сантиметров до 1 метра. Поскольку внешнее полиимидное покрытие непрозрачно в УФ-области, то участок покрытия удаляют и создают окно для УФ-детектирования. Капилляр закрепляется в специальной пластиковой кассете.
Надежное термостатирование капилляра является основным условием получения воспроизводимых времен миграции определяемого соединения и площади результирующего пика, что важно для количественного анализа. Используют капилляры с внутренним диаметром 25-50 мкм, что является компромиссным решением между достаточно высокой чувствительностью и эффективностью разделения.
Надежное термостатирование капилляра является основным условием получения воспроизводимых времен миграции определяемого соединения и площади результирующего пика, что важно для количественного анализа. Используют капилляры с внутренним диаметром 25-50 мкм, что является компромиссным решением между достаточно высокой чувствительностью и эффективностью разделения.

Слайд 52Ввод образца
Проба может быть введена в капилляр электрофоретическим, электрокинетическим или вытеснительным
способом. Объем вводимой пробы не превышает 2 нл, относительное стандартное отклонение составляет 0,03-0,04. При электрофоретическом вводе пробы, к концам капилляра прикладывается высокое напряжение на фиксированный промежуток времени, при этом входной конец капилляра погружают в раствор пробы. Ионы пробы перемещаются в капилляр пропорционально их электрофоретической подвижности. В случае электрокинетического ввода, компоненты пробы попадают в капилляр за счет комбинации электроэндоосмотического давления и электрофоретической подвижности. Вытеснительный ввод пробы достигается либо за счет создания избыточного внешнего давления инертного газа, приложенного к резервуару с образцом, либо за счет создания вакуума на выходе из капилляра или путем изменения уровня/высоты резервуара, содержащего образец, относительно резервуара с буферным раствором на выходе из капилляра (так называемое гравитационное введение пробы).


Слайд 54Пример разделения неорганических ионов (КЭ)
1 – хлорид; 2 – нитрит; 3
–
сульфат; 4 – нитрат; 5 – фторид; 6 – гидрофосфат; 7 – гидрокарбонат;
сульфат; 4 – нитрат; 5 – фторид; 6 – гидрофосфат; 7 – гидрокарбонат;
1 – хлорид; 2 – нитрит; 3 –сульфат; 4 – нитрат;")

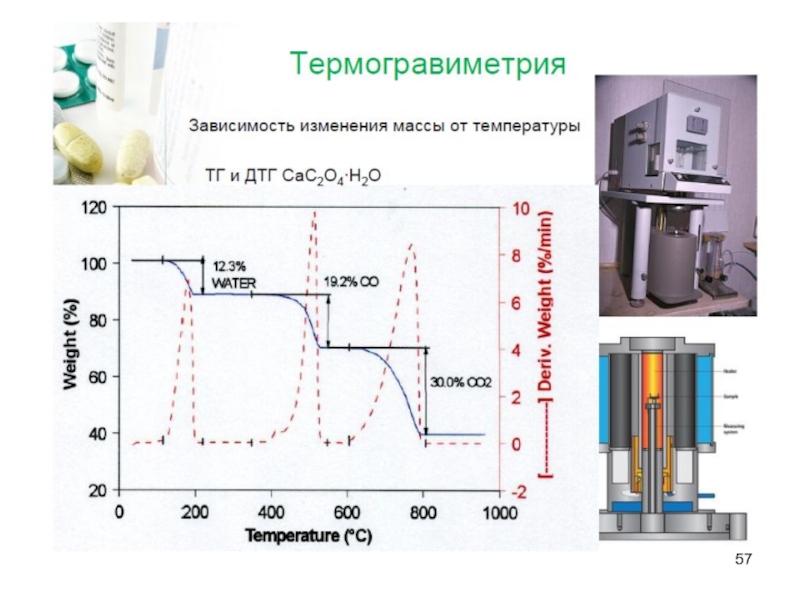
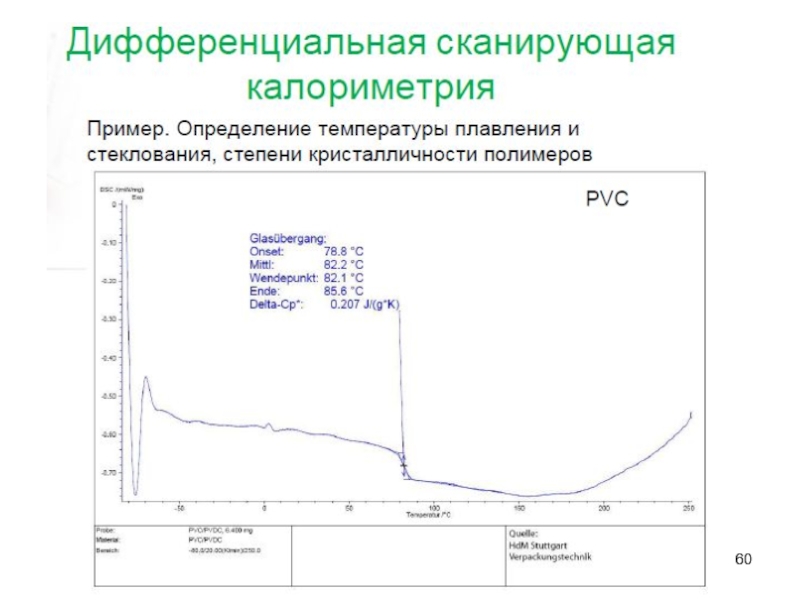
Слайд 56Термические методы анализа
Основаны на установлении зависимостей различных физических или физико-химических свойств
веществ от температуры (градиента температуры).
А – Термогравиметрия
Б – Дифференциальный термический анализ
В – Дифференциальная сканирующая калориметрия
А – Термогравиметрия
Б – Дифференциальный термический анализ
В – Дифференциальная сканирующая калориметрия