ДНК:
1 5 10
tagcaaaatg
- Главная
- Разное
- Дизайн
- Бизнес и предпринимательство
- Аналитика
- Образование
- Развлечения
- Красота и здоровье
- Финансы
- Государство
- Путешествия
- Спорт
- Недвижимость
- Армия
- Графика
- Культурология
- Еда и кулинария
- Лингвистика
- Английский язык
- Астрономия
- Алгебра
- Биология
- География
- Детские презентации
- Информатика
- История
- Литература
- Маркетинг
- Математика
- Медицина
- Менеджмент
- Музыка
- МХК
- Немецкий язык
- ОБЖ
- Обществознание
- Окружающий мир
- Педагогика
- Русский язык
- Технология
- Физика
- Философия
- Химия
- Шаблоны, картинки для презентаций
- Экология
- Экономика
- Юриспруденция
Модели молекулярной эволюции, кладистика по Хеннигу и метод максимальной парсимонии презентация
Содержание
- 1. Модели молекулярной эволюции, кладистика по Хеннигу и метод максимальной парсимонии
- 2. Модели молекулярной эволюции ДНК:
- 3. Соотношения между нуклеотидными заменами и нуклеотидными
- 4. Единичная замена Множественные замены Параллельные замены Конвергентные
- 5. явные и скрытые генетические дистанции 1 10
- 6. Единичная замена Множественные замены Число нуклеотидных замен
- 7. Наблюдаемые генетические дистанции как правило меньше
- 8. Закономерности накопления замен
- 9. ACGTACGTAC CCGTACGTAC ACGTACGTAC Первая замена - в
- 10. CCGAACGTAC ACGTACGTAC Вторая замена – Имеется
- 11. CGTACGTACG ACGTACGTAC Третья замена имеет большую вероятность
- 12. Зависимость между временем дивергенции и числом наблюдаемых
- 13. “Сырые” (нескорректированные) генетические дистанции легко вычислить,
- 14. Purines = adenin and guanine Pirimidines = cytosine and thymine
- 15. Кривые накопления повторных замен для транзиций и
- 16. Кривая накопления транзиций по отношению к трансверсиям
- 17. Генетический код Замена в первой позиции кодона
- 18. Кривые накопления повторных замен для третьей и первой позиций кодона
- 19. Какие параметры можно извлечь из нуклеотидного выравнивания? 1 10 20 30
- 20. Какие параметры можно извлечь из нуклеотидного выравнивания?
- 21. Какие параметры можно извлечь из нуклеотидного выравнивания?
- 22. Какие параметры можно извлечь из нуклеотидного выравнивания?
- 23. Какие параметры можно извлечь из нуклеотидного выравнивания?
- 24. Какие параметры можно извлечь из нуклеотидного выравнивания?
- 25. Какие параметры можно извлечь из нуклеотидного выравнивания?
- 26. Какие параметры можно извлечь из нуклеотидного выравнивания?
- 27. Какие параметры можно извлечь из нуклеотидного выравнивания?
- 28. Предпосылки 1) нуклеотидные замены одного типа равновероятны
- 29. Если вероятности нуклеотидных замен (p) и частоты
- 30. Если вероятности нуклеотидных замен (p) и частоты
- 31. частоты нуклеотидов и доли замен разного типа берутся непосредственно из выравнивания
- 32. JC Вероятности всех замен одинаковы, частоты нуклеотидов
- 33. Двухпараметрическая модель Кимуры K2P Вероятности транзиций и
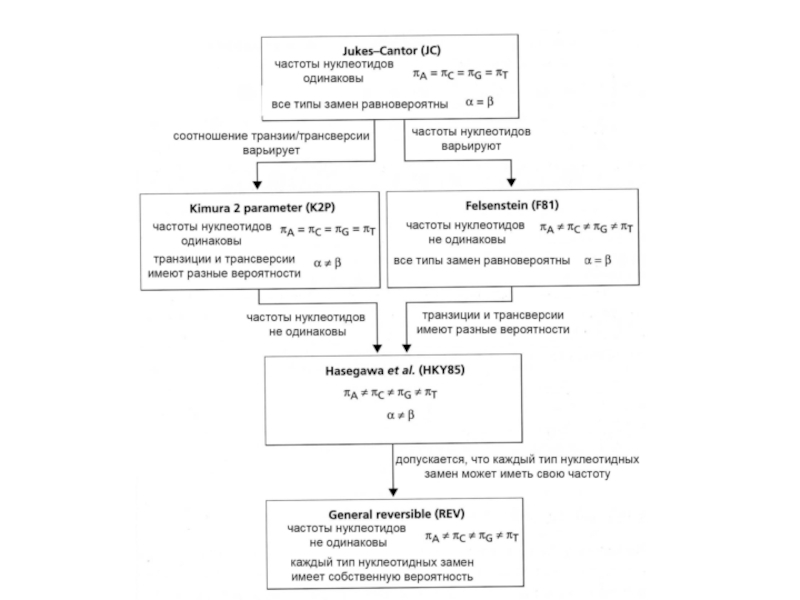
- 34. F81 Вероятности всех замен одинаковы, но частоты нуклеотидов разные
- 35. HKY model Вероятности транзиций и трансверсий разные, частоты нуклеотидов разные
- 36. REV Вероятности ВСЕХ ЗАМЕН разные, частоты нуклеотидов разные
- 38. Чем хороши и чем плохи сложные и простые модели?
- 39. Условия, при которых работают эти модели Это
- 40. Структура 18S rDNA
- 41. 18S rDNA (фрагмент) 18S rDNA (фрагмент)
- 42. Условия, при которых работают эти модели Все
- 43. Общие принципы построения филогений
- 44. Подходы к выявлению филогений традиционный (Геккелевский, эмпирико-интуитивный)
- 45. Традиционный (эмпирико-интуитивный) метод выведения филогений Строго
- 46. По Геккелю филогенетика – наука о путях,
- 47. Н.Я.Кузнецов. Насекомые чешуекрылые. Т. 1. Фауна России.
- 48. “Недавно в лабораторию [Моргана]пришла почта с произведениями
- 49. Традиционная кладистика (Hennig, 1950, 1966)
- 50. Признаки Негомологичные (гомоплазии) Гомологичные Плезиоморфии Апоморфии Синапоморфии
- 51. Гомоплазии – независимо возникшие признаки. Они не несут никакой информации о филогении 1 - гомоплазия
- 52. Плезиоморфии – древние (=исходные; =примитивные) гомологичные признаки.
- 53. Апоморфия – новый (=продвинутый; =производный; =прогрессивынй)
- 54. Но если апоморфия возникла до разделения ветвей
- 55. Для построения филогении трех таксонов (два ветвления)
- 56. В общем виде для полного разрешения филогении,
- 57. Филогения строится как система соподчиненных (вложенных
- 58. Модель эволюции в кладистике по Геннигу Топология - строгая дихотомия Процесс – накопление синапоморфий.
- 59. Алгоритм анализа Одна истинная синапоморфия может разрешить
- 60. Построение молекулярного дерева с использованием кладистики по
- 61. Построение молекулярного дерева таксонов 1-4 с использованием
- 62. Состояние ACGTA плезиоморфно 1 AAGTT
- 63. A во второй позиции – синапоморфия 1
- 64. T в пятой позиции – синапоморфия
- 65. Проблема гомоплазий Презумпция: Синапоморфии встречаются чаще, чем гомоплазии
- 66. Конфликт между потенциальными синапоморфиями 1 AAGTT
- 67. Принципы традиционной кладистики Если возникает конфликт между
- 68. Другие проблемы генниговской кладистики: “Надежных” синапоморфий может
- 69. Проблемы традиционной кладистики “Надежных” синапоморфий может быть
- 70. Проблемы традиционной кладистики “Надежных” синапоморфий может быть
- 71. Картины филогенезов, которуе создает кладистический (по Геннигу
- 72. Принцип монофилии лежит в самой основе
- 73. Кладизм объявляет парафилетические группы вне закона просто
- 74. Проблемы парафилетических таксонов 1+2 = парафилетический таксон.
- 75. Монофилетический таксон - группа, которая включает предка
- 76. Перипатрическое видообразование: предковый таксон при этом не
- 77. Филогеография медведей, основанная на кладистическом анализе (MP) нуклеотидных замен в митохондриальном геноме (Avise, 2004)
- 78. Кладистика по Геннигу остается рабочим инструментом филогенетики!
- 79. Фенетика В кладистике процедура выявления гомологичных признаков
- 80. Фенетика Отказ от доминирования принципа гомологии (в
- 81. Фенетика Кластерный анализ (выявление группировок по
- 82. Фенетика Пример научной, но неправильной (неадекватной)
- 83. Традиционная и нумерическая кладистика Увеличение числа
- 84. Если возникает конфликт между потенциальными синапоморфиями, то
- 85. Если возникает конфликт между потенциальными синапоморфиями, то
- 86. Нумерическая кладистика и метод максимальной парсимонии
- 87. Метод максимальной парсимонии (наибольшей экономии)
- 88. Нет гомоплазий – одно возможное дерево Число
- 89. Первое дерево более парсимониальное, оно короче Происходит голосование “синапоморфиями”
- 90. в реальности у нас исходно нет
- 91. Шаг 1: выявление признаков и их состояний
- 92. Признак – цвет глаз Состояния –
- 93. Шаг 3: Составление матрицы признаков
- 94. Бинарная матрица Матрица множественных состояний
- 95. Нуклеотидное (или аминокислотное) выравнивание – это
- 96. Шаг 4: выбор модели эволюции Модель Камина-Сокола
- 97. Модель Долло (Dollo parsimony) (основана на принципе
- 98. Модель Фитча-Вагнера (Fitch-Wagner parsimony) – симметричная модель 0
- 99. Модель Фитча-Вагнера (Fitch-Wagner parsimony) для множественных состояний признака 0
- 100. Модель Фитча-Вагнера (Fitch-Wagner parsimony) для нуклеотидных замен A
Слайд 1Лекция 4 Модели молекулярной эволюции, кладистика по Хеннигу и метод максимальной
парсимонии


Слайд 3Соотношения между нуклеотидными
заменами и нуклеотидными различиями
Единичная замена
Множественные замены
Параллельные замены
Конвергентные замены
Обратная
замена
Одновременные замены
в разных линиях

Слайд 4Единичная замена
Множественные замены
Параллельные замены
Конвергентные замены
Обратная замена
Одновременные замены
в разных линиях
Число нуклеотидных замен
≥ числа наблюдаемых нуклеотидных различий

Слайд 5явные и скрытые генетические дистанции
1
10
20
30
Что такое генетическая дистанция?
d = p, где
p – доля различающихся сайтов
d – это “сырая” дистанция
d – это “сырая” дистанция
d = 3/32=9.375%

Слайд 6Единичная замена
Множественные замены
Число нуклеотидных замен ≥ числа наблюдаемых нуклеотидных отличий
Проблема дистанций
состоит в том, что наблюдаемые дистанции
могут быть меньше, чем реальные дистанции, так как не все
замены видны при сравнении сиквенсов
могут быть меньше, чем реальные дистанции, так как не все
замены видны при сравнении сиквенсов

Слайд 7
Наблюдаемые генетические дистанции как правило меньше реальных эволюционных дистанций, так как
есть скрытые замены
Но как выявить эти реальные эволюционные дистанции?
Нужно знать возраст таксонов (время дивергенции) и скорость замен
Но как выявить эти реальные эволюционные дистанции?
Нужно знать возраст таксонов (время дивергенции) и скорость замен


Слайд 9ACGTACGTAC
CCGTACGTAC
ACGTACGTAC
Первая замена - в сайте 1. d=0.1
Наблюдаемая дистанция = реальной
дистанции

Слайд 10CCGAACGTAC
ACGTACGTAC
Вторая замена –
Имеется вероятность 0.1, что она будет повторной
(т.е.
тоже в сайте 1) и вероятность 0.9, что она будет неповторной
Если она все же будет в первой позиции, то
Наблюдаемая дистанция = 0.1 (или даже 0),
а истинная дистанция = 0.2
Если она все же будет в первой позиции, то
Наблюдаемая дистанция = 0.1 (или даже 0),
а истинная дистанция = 0.2
Но скорее всего (с вероятностью 0.9), вторая замена не будет в сайте 1
1 2 3 4 5 6 7 8 9 10
")
Слайд 11CGTACGTACG
ACGTACGTAC
Третья замена имеет большую вероятность быть повторной,
четвертая – еще большую,
и. т.д.
Т.е. чем больше замен, тем больше вероятность повторных замен.
Если все 10 позиций испытали замены,
то любая следующая замена будет повторной.
Т.е. чем больше замен, тем больше вероятность повторных замен.
Если все 10 позиций испытали замены,
то любая следующая замена будет повторной.
После этого замены продолжают накапливаться,
а наблюдаемые различия не растут

Слайд 12Зависимость между временем дивергенции и числом наблюдаемых
нуклеотидных отличий в гене
CytB у жвачных копытных животных
Происходит насыщение нуклеотидными заменами –
число замен растет, но уровень отличий выходит
на плато и не меняется
NB: каждая точка – это пара особей разных линий

Слайд 13“Сырые” (нескорректированные)
генетические дистанции легко
вычислить, но они могут быть
сильно занижены.
Необходима
коррекция
Ее можно сделать с
использованием моделей,
которые учитывают разницу
в эволюции разных признаков
Ее можно сделать с
использованием моделей,
которые учитывают разницу
в эволюции разных признаков
генетические дистанции легко вычислить, но они могут быть сильно занижены.Необходима коррекцияЕе можно сделать с")

Слайд 15Кривые накопления повторных замен для транзиций и трансверсий
Каждая точка –
это сравнение,
т.е.
пара видов


Слайд 17Генетический код
Замена в первой позиции кодона ведет к замене аминокислоты
Замена в
третьей позиции кодона как правило синонимична
—› нуклеотиды в третьей позиции эволюционируют быстрее
—› нуклеотиды в третьей позиции эволюционируют быстрее



Слайд 20Какие параметры можно извлечь из нуклеотидного выравнивания?
(1) Длина
(2) Доля изменчивых сайтов
(3) Доля инвариантных сайтов
(4) Соотношение нуклеотидов разных типов
(5) Доля транзиций и трансверсий
(6) Доля нуклеотидных замен разных типов
(7) Все это (2-6) отдельно для каждой позиции кодона
(8) Доля синонимичных и несинонимичных замен
(9) Доля синонимичных и несинонимичных замен
отдельно для каждой позиции кодона
(10) Распределение замен по длине нуклеотидной последовательности
1
10
20
30
 Длина(2) Доля изменчивых сайтов (3) Доля инвариантных сайтов(4)")
Слайд 21Какие параметры можно извлечь из нуклеотидного выравнивания?
Длина выравнивания
Зависит от задач и
технических возможностей
1
10
20
30

Слайд 22Какие параметры можно извлечь из нуклеотидного выравнивания?
(2) Доля изменчивых сайтов
(3)
Доля инвариантных сайтов
1
10
20
30
 Доля изменчивых сайтов (3) Доля инвариантных сайтов1102030")
Слайд 23Какие параметры можно извлечь из нуклеотидного выравнивания?
(4) Соотношение нуклеотидов разных типов
(A C G T)
1
10
20
30
 Соотношение нуклеотидов разных типов (A C G T)1102030")
Слайд 24Какие параметры можно извлечь из нуклеотидного выравнивания?
(5) Доля транзиций и трансверсий
1
10
20
30
 Доля транзиций и трансверсий1102030")
Слайд 25Какие параметры можно извлечь из нуклеотидного выравнивания?
(6) Доля нуклеотидных замен разных
типов
1
10
20
30
 Доля нуклеотидных замен разных типов1102030")
Слайд 26Какие параметры можно извлечь из нуклеотидного выравнивания?
(7) Все это (2-6) отдельно
для каждой позиции кодона
1
10
20
30
 Все это (2-6) отдельно для каждой позиции кодона1102030")
Слайд 27Какие параметры можно извлечь из нуклеотидного выравнивания?
(8) Доля синонимичных и несинонимичных
замен
(9) Доля синонимичных и несинонимичных замен
отдельно для каждой позиции кодона
(10) Распределение замен по длине нуклеотидной последовательности
(9) Доля синонимичных и несинонимичных замен
отдельно для каждой позиции кодона
(10) Распределение замен по длине нуклеотидной последовательности
1
10
20
30
 Доля синонимичных и несинонимичных замен(9) Доля синонимичных и")
Слайд 28Предпосылки
1) нуклеотидные замены одного типа равновероятны в разных частях одного гена
2)
нуклеотидные замены обратимы (здесь не работает принцип необратимости эволюции) (A <-> T)
Модели нуклеотидных замен
 нуклеотидные замены одного типа равновероятны в разных частях одного гена2) нуклеотидные замены обратимы (здесь")
Слайд 29Если вероятности нуклеотидных замен (p) и частоты нуклеотидов (f) константны во
времени, то суммарная эволюционная дистанция ( доля измененных нуклеотидов) =
Где t это время, PAC –
PAC = PCA
 и частоты нуклеотидов (f) константны во времени, то суммарная эволюционная")
Слайд 30Если вероятности нуклеотидных замен (p) и частоты нуклеотидов (f) константны во
времени, то суммарная эволюционная дистанция ( доля измененных нуклеотидов) =
 и частоты нуклеотидов (f) константны во времени, то суммарная эволюционная")

Слайд 32JC
Вероятности всех замен одинаковы, частоты нуклеотидов равны
D=
D = -(3/4)ln(1-4/3
где p –
это сырая дистанция
ln(1-4/3где p – это сырая дистанция")
Слайд 33Двухпараметрическая модель Кимуры K2P
Вероятности транзиций и трансверсий разные,
частоты нуклеотидов равны
α
– транзиция
β - трансверсия
β - трансверсия





Слайд 39Условия, при которых работают эти модели
Это стохастические модели, которые предполагают, что
все замены случайны и независимы друг от друга
А если нет …
А если нет …


18S rDNA(фрагмент)")
Слайд 42Условия, при которых работают эти модели
Все замены случайны и независимы друг
от друга
А если нет …
Зависимые компенсаторные замены
А если нет …
Зависимые компенсаторные замены

Слайд 43
Общие принципы построения филогений
1) Анализ признаков,
2) выбор оптимальной
модели эволюции признака,
3) выбор методов и алгоритмов для построения дерева
3) выбор методов и алгоритмов для построения дерева
 Анализ признаков, 2) выбор оптимальной модели эволюции признака, 3)")
Слайд 44Подходы к выявлению филогений
традиционный (Геккелевский, эмпирико-интуитивный)
традиционная кладистика (Hennig, 1950, 1966)
фенетика
метод максимальной парсимонии
метод максимального правдоподобия
метод Байеса
методы, основанные на анализе генетических дистанций
 традиционная кладистика (Hennig, 1950, 1966) фенетикаметод максимальной парсимонииметод максимального")
Слайд 45Традиционный (эмпирико-интуитивный) метод выведения филогений
Строго научный и, как правило, очень качественный
анализ признаков сочетается с частично или полностью интуитивным методом их филогенетического обобщения,
то есть с отсутствием универсальных, четких и формализованных алгоритмов филогенетического анализа
Модели эволюции примитивны и не формализованы
то есть с отсутствием универсальных, четких и формализованных алгоритмов филогенетического анализа
Модели эволюции примитивны и не формализованы
 метод выведения филогений Строго научный и, как правило, очень качественный анализ признаков сочетается")
Слайд 46По Геккелю филогенетика – наука о путях,
закономерностях и причинах исторического
развития организмов
Ernst Haeckel (1834-1919)

Слайд 47Н.Я.Кузнецов. Насекомые чешуекрылые. Т. 1. Фауна России. Петроград, 1915
Однако обоснование филогений
ограничивается словами: «Я предлагаю принять филогенетические отношения, представленные на рисунках»
великолепный анализ морфологии
Выявление гомологий

Слайд 48 “Недавно в лабораторию [Моргана]пришла почта с произведениями Северцова с многочисленными филогенетическими
древесами, на которые я указал Моргану. Его реплика была такова: “Я думал, что такие идиоты могут существовать только в Museum of Natural History”. После этого я со сладострастием наблюдал, как все это пошло на свалку”
Ф.Г. Добржанский (из письма к Ю.А.Филипченко, 23 июля 1928)
Ф.Г. Добржанский (из письма к Ю.А.Филипченко, 23 июля 1928)
Ф.Г.Добржанский
фото 1935 г.

Слайд 49 Традиционная кладистика (Hennig, 1950, 1966) Хенниг предложил строго научные принципы перехода от
анализа признаков к реконструкции филогений
Willi Hennig
(1913-1976)
 Хенниг предложил строго научные принципы перехода от анализа")
Гомологичные ПлезиоморфииАпоморфииСинапоморфии")
Слайд 51Гомоплазии – независимо возникшие признаки. Они не несут никакой информации о
филогении
1 - гомоплазия

Слайд 52Плезиоморфии – древние (=исходные; =примитивные) гомологичные признаки. Они не несут никакой
информации о топологии поздних ветвлений.
 гомологичные признаки. Они не несут никакой информации о топологии поздних ветвлений.")
Слайд 53Апоморфия – новый
(=продвинутый; =производный; =прогрессивынй)
гомологичный признак.
Единичная апоморфия, возникшая
в концевой ветви, метит только эту ветвь
и не несет никакой информации о топологии
и не несет никакой информации о топологии
Апоморфия является специфическим маркером эволюционной линии
 гомологичный признак. Единичная апоморфия, возникшая в концевой ветви,")
Слайд 54Но если апоморфия возникла до разделения ветвей и передалась в обе
ветки, то наличие такой апоморфии указывает на существование клады, состоящей из двух таксонов.
Такая апоморфия называется синапоморфией.
Синапоморфия несет информацию о филогении!!!
Такая апоморфия называется синапоморфией.
Синапоморфия несет информацию о филогении!!!

Слайд 55Для построения филогении трех таксонов (два ветвления) необходимо наличие одной синапоморфии
Для
построения филогении трех таксонов (два ветвления) необходимо наличие одной синапоморфии
 необходимо наличие одной синапоморфииДля построения филогении трех таксонов")
Слайд 56В общем виде для полного разрешения филогении, включающей n ветвлений, необходимо
и достаточно n-1 синапоморфий (по одной на каждый узел, кроме базального)
В общем виде для полного разрешения филогении, включающей n ветвлений, необходимо и достаточно n-1 синапоморфий (по одной на каждый узел, кроме базального)

Слайд 57
Филогения строится как система соподчиненных (вложенных одна в другую) клад (монофилетических
групп), каждая из которых выявляется по наличию синапоморфий
 клад (монофилетических групп), каждая из которых")
Слайд 58Модель эволюции в кладистике по Геннигу
Топология - строгая дихотомия
Процесс – накопление
синапоморфий.

Слайд 59Алгоритм анализа
Одна истинная синапоморфия может разрешить узел ветвления филогенетического дерева
Выявление филогении
– многоступенчатый процесс выдвижения и тестирования филогенетических гипотез, в ходе которого представление о филогенезе постепенно уточняется и конкретизируется


Слайд 61Построение молекулярного дерева таксонов 1-4 с использованием кладистики по Хеннигу
1
AAGTT
2 AAGTT
3 ACGTT
4 ACGTA
5 ACGTA
6 ACGTA
7 ACGTA
2 AAGTT
3 ACGTT
4 ACGTA
5 ACGTA
6 ACGTA
7 ACGTA






Слайд 67Принципы традиционной кладистики
Если возникает конфликт между потенциальными синапоморфиями, то основной путь
его решения – переисследование материала, поиск и изучение дополнительных признаков и таксонов

Слайд 68Другие проблемы генниговской кладистики:
“Надежных” синапоморфий может быть мало, недостаточно для того,
что разрешить все узлы ветвления разрабатываемой филогении

Слайд 69Проблемы традиционной кладистики
“Надежных” синапоморфий может быть мало, недостаточно для того, что
разрешить все узлы ветвления разрабатываемой филогении
отбрасывая «ненадежные» признаки, мы теряем филогенетическую информацию, так как «ненадежные» признаки также могут содержать филогенетический сигнал
отбрасывая «ненадежные» признаки, мы теряем филогенетическую информацию, так как «ненадежные» признаки также могут содержать филогенетический сигнал

Слайд 70Проблемы традиционной кладистики
“Надежных” синапоморфий может быть мало, недостаточно для того, что
разрешить все узлы ветвления разрабатываемой филогении
отбрасывая «ненадежные» признаки, мы теряем филогенетическую информацию, так как «ненадежные» признаки также могут содержать филогенетический сигнал
Реконструируется только топология!
отбрасывая «ненадежные» признаки, мы теряем филогенетическую информацию, так как «ненадежные» признаки также могут содержать филогенетический сигнал
Реконструируется только топология!

Слайд 71Картины филогенезов, которуе создает кладистический (по Геннигу и парсимониальный) анализ, неполны
и однобоки:
Анагенез не учитывается
Ретикулогенез (слияния+интрогрессии) не выявляется
Некоторые узлы принципиально не могут быть выявлены
 анализ, неполны и однобоки: Анагенез не")
Слайд 72Принцип монофилии лежит
в самой основе алгоритма
построения дерева в
хенниговской
кладистике.
Сипапоморфии однозначно
определяют только
монофилетические линии,
а немонофилетические
группы, например,
парафилетические
группировки
не могут быть
определены однозначно.
Сипапоморфии однозначно
определяют только
монофилетические линии,
а немонофилетические
группы, например,
парафилетические
группировки
не могут быть
определены однозначно.

Слайд 73Кладизм объявляет парафилетические группы вне закона просто по той причине, что
он не умеет их выявлять
(поскольку парафилетические группы не имеют синапоморфий)

Слайд 74Проблемы парафилетических таксонов
1+2 = парафилетический таксон. Признак A не уникален, признак
B характеризует лишь часть таксона 1+2 и тоже не уникален
1+3 = парафилетический таксон. Признак A не уникален, признак B характеризует лишь часть таксона 1+3 и тоже не уникален
Существует несколько вариантов частично
пересекающихся парафилетических таксонов

Слайд 75Монофилетический таксон - группа, которая включает предка и всех его потомков
Монофилетические группы могут иметь синапоморфии
A – это синапоморфия таксона 1+(2+3)
→ A однозначно характеризует таксон 1+(2+3)
B, синапоморфия таксона 2+3
→ B однозначно характеризует таксон 2+3
Другие варианты монофилетических таксонов не существуют

Слайд 76Перипатрическое видообразование: предковый таксон при этом не исчезает, но он становится
парафилетическим.
Несмотря на парафилию, такой вид представляет собой единое репродуктивное сообщество, изолированное от дочерних видов

Слайд 77Филогеография медведей, основанная на кладистическом анализе (MP) нуклеотидных замен в митохондриальном
геноме (Avise, 2004)
 нуклеотидных замен в митохондриальном геноме (Avise, 2004)")

Слайд 79Фенетика
В кладистике процедура выявления гомологичных признаков (дифференциация от гомоплазий) не формализована.
Это может быть причиной субъективизма
 не формализована. Это может быть причиной субъективизма")
Слайд 80Фенетика
Отказ от доминирования принципа гомологии (в фенетике все признаки имеют равный
вес)
Степень родства = степени сходства
+ попытка ввести объективность в систематику и филогенетику
+ широкое внедрение методов статистики в систематику
Степень родства = степени сходства
+ попытка ввести объективность в систематику и филогенетику
+ широкое внедрение методов статистики в систематику
Степень родства = степени")
Слайд 81Фенетика
Кластерный анализ (выявление группировок по степени их сходства).
Иерархии таких группировок
можно интерпретировать в качестве филогении.
.Иерархии таких группировок можно интерпретировать в качестве")
Слайд 82Фенетика
Пример научной, но неправильной (неадекватной) методологии
Научность – строгое следование принципам
научной логики, избегание субъективизма
Неправильность – основана на неадекватной аксиоматике (на ложных предпосылках)
Неправильность – основана на неадекватной аксиоматике (на ложных предпосылках)
 методологииНаучность – строгое следование принципам научной логики, избегание субъективизмаНеправильность")
Слайд 83Традиционная и нумерическая кладистика
Увеличение числа признаков приводит к противоречиям между предполагаемыми
синапоморфиями, которые свидетельствуют о наличии гомоплазий
При наличии противоречий между “синапоморфиями” возможны разные варианты филогении
Как выбрать правильный вариант?
При наличии противоречий между “синапоморфиями” возможны разные варианты филогении
Как выбрать правильный вариант?

Слайд 84Если возникает конфликт между потенциальными синапоморфиями, то есть два пути его
решения:
1)переисследование материала, поиск и изучение дополнительных признаков и таксонов с целью выявления “истинных” синапоморфий - Традиционная кладистика
1)переисследование материала, поиск и изучение дополнительных признаков и таксонов с целью выявления “истинных” синапоморфий - Традиционная кладистика
переисследование материала, поиск")
Слайд 85Если возникает конфликт между потенциальными синапоморфиями, то есть два пути его
решения:
1)переисследование материала, поиск и изучение дополнительных признаков и таксонов с целью выявления “истинных” синапоморфий
2) наоборот - использование большого числа признаков, получение нескольких (многих) деревьев и выбор “лучшего” из них c использованием определенного критерия -нумерическая кладистика
1)переисследование материала, поиск и изучение дополнительных признаков и таксонов с целью выявления “истинных” синапоморфий
2) наоборот - использование большого числа признаков, получение нескольких (многих) деревьев и выбор “лучшего” из них c использованием определенного критерия -нумерическая кладистика
переисследование материала, поиск")
Слайд 86Нумерическая кладистика и метод максимальной парсимонии
При наличии противоречий между “синапоморфиями” возможны
разные варианты филогении
Как выбрать “правильное” дерево?
- критерий максимальной парсимонии
Как выбрать “правильное” дерево?
- критерий максимальной парсимонии

")
Слайд 88Нет гомоплазий – одно возможное дерево
Число шагов (L) = 3
Сай4 –
инвариантный, сайт 3 - вариабельный
 = 3Сай4 – инвариантный, сайт 3 - вариабельный")

Слайд 90 в реальности у нас исходно нет ни топологии дерева, ни
распределения признаков по нему, ни анцестрального состояния.
Как все это найти?
Как все это найти?

Слайд 91Шаг 1: выявление признаков и их состояний
Признак – цвет глаз
Состояния –
коричневый, голубой, зеленый
Признак – группа крови
Состояния – первая, вторая, третья, четвертая

Слайд 92Признак – цвет глаз
Состояния – коричневый (0), голубой (1), зеленый (2)
Признак
– группа крови
Состояния – первая (0), вторая (1), третья (2), четвертая (3)
Состояния – первая (0), вторая (1), третья (2), четвертая (3)
Шаг 2: кодирование признаков и их состояний
0 – обычно анцестральное состояние
, голубой (1), зеленый (2)Признак – группа кровиСостояния –")


Слайд 95 Нуклеотидное (или аминокислотное) выравнивание – это уже готовая матрица признаков
4
состояния – A C G T
 выравнивание – это уже готовая матрица признаков4 состояния – A C")
Слайд 96Шаг 4: выбор модели эволюции
Модель Камина-Сокола (Camin- Sokal parsimony): анцестральное состояние
известно, тогда 0 —› 1
Всегда дает укорененное дерево
Всегда дает укорененное дерево
: анцестральное состояние известно, тогда 0 —›")
Слайд 97Модель Долло (Dollo parsimony) (основана на принципе необратимости эволюции) - допускаются
изменения признака в любую сторону, но только один раз (вернее повторные изменения менее вероятны)
 (основана на принципе необратимости эволюции) - допускаются изменения признака в любую")
Слайд 98 Модель Фитча-Вагнера (Fitch-Wagner parsimony) – симметричная модель
0
дерево неукорененное!!!
 – симметричная модель 0")
Слайд 99 Модель Фитча-Вагнера (Fitch-Wagner parsimony) для множественных состояний признака
0
0 <—› 2
0 <—› 3
1 <—› 2
1 <—› 3
2 <—› 3
дерево неукорененное!!!
 для множественных состояний признака 0")
Слайд 100 Модель Фитча-Вагнера (Fitch-Wagner parsimony) для нуклеотидных замен
A
A <—› G A <—› T
C <—› G C <—› T
G <—› T
C <—› G C <—› T
G <—› T
 для нуклеотидных замен A")





